The described protocol is a straightforward approach to isolate neurons, macrophages and microglia from zebrafish larval brains. From these isolated cells, significant amounts of high quality (RIN > 7) RNA were extracted. The aim of this protocol is to isolate different types of cells from the CNS, with minimal modification of their gene expression profile to analyze and characterize cell properties and functions. Therefore, the entire protocol is performed at 4 °C with a mechanical brain tissue homogenization. This method has been successfully used for two studies performed in the laboratory. In the first study, neurons and macrophages/microglia were isolated from 8 dpf mpeg1:eGFP+/NBT:DsRed+ larvae (Figure 1). FACS allowed cell separation from debris in function of their size (FSC-A) and granularity (SSC-A) (Figure 1A). Single cells were then separated from doublets or cell agglomerates (Figure 1B). From the single cell population, a gate was drawn to eliminate dead cells (DAPI+). The corresponding dot plot revealed that this experimental protocol preserves cell plasma membrane integrity, as the rate of dead cells is only 26.7% (Figure 1C). Finally, neurons (DsRed+) and macrophages/microglia (GFP+) were easily segregated from the live cell population gates. The neuron population (23.1 %) appeared to be more prominent than the macrophages/microglia population (1.56 %) within the brain (Figure 1D). This protocol has allowed to isolate RNA from those cells to perform subsequent qPCR analyses to compare the expression of specific genes between neurons and macrophages/microglia. Figure 2 shows neuronal and macrophages/microglia gene expression levels of proliferating cell nuclear antigen (pcna)against the β-actin house-keeping gene as an example.
For the second study this method focused on microglia isolation from 3, 5 and 7 dpf larval brains. In contrast to the experiment described above, cells were isolated by immuno-staining using 4C4, an antibody which specifically labels microglia (Figure 3 A-D). As previously described, microglia (4C4+) were selected from live cells and collected (Figure 3D). Microglia numbers within zebrafish larval brains are variable (Table 1), and very low at 3 dpf (∼ 25 per fish). Quality and quantity of extracted RNA from those cells were measured using a micro-capillary electrophoresis based system. Results obtained of extracted RNA from microglia of 5 dpf larval zebrafish brains have been provided to illustrate an example of RNA analysis (Table 1 (5 dpf; experiment 4)). Figure 4 shows the electrophoresis trace and its graphic representation obtained for this sample with a clear visualization of ribosomal RNA (28s and 18s). This data is necessary to calculate sample RIN and to determine RNA concentration. Table 1 summarizes the number of isolated microglia per fish, the amount of RNA per microglia and the RIN score obtained for each different experiment at 3, 5 and 7 dpf. The amount and the quality of extracted RNA from isolated microglia using this method allowed us to amplify the RNA into cDNA using a kit. Quality and quantity tests provided by Edinburgh Genomics confirm that the amplified cDNA is of sufficient quality for library preparation and subsequent sequencing. Figure 5 shows the size distribution of cDNA fragments and their amount measured using an electrophoretic system. In this sample, the cDNA had a mean size of 299bp at a concentration of 36100 pmol/l. Table 2 illustrates respectively quality and quantity tests made on amplified cDNA from RNA samples (Table 1 (5 dpf; experiment 4)). The amplified cDNA has been used successfully for sequencing.
Several studies performed in the laboratory confirmed that the quality and quantity of extracted RNA from neurons, macrophages and microglia can be used for subsequent qPCR and genome wide gene expression analyses. Therefore, this experimental protocol can be used to reliably isolate different types of CNS cells without altering their membrane integrity and limiting modification of their gene expression profile.
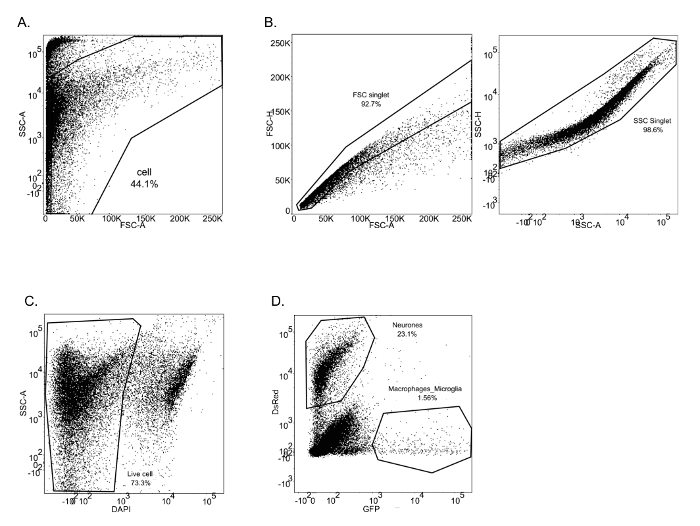
Figure 1: FACS sorting for neurons and macrophages/microglia from mpeg1:GFP+/NBT:DsRed+ 8 dpf zebrafish larvae. (A-C) Successive gating shows sequential selection of all brain cells (A), single cells by forward scatter and side scatter (B). (C) Dead cells were excluded by DAPI labelling. (D) Neurons and macrophages/microglia were identified respectively by DsRed and GFP positive staining. Please click here to view a larger version of this figure.
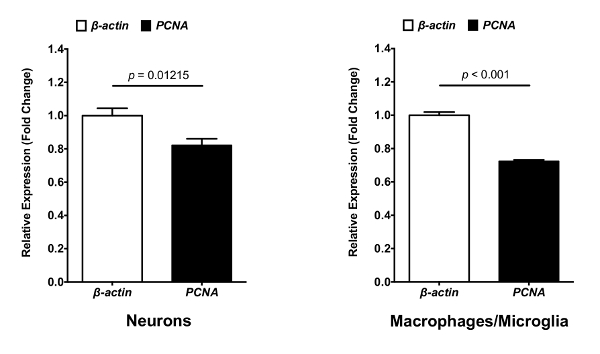
Figure 2: Gene expression analysis for pcna and β-actin in neurons and macrophages/microglia. RNA from isolated neurons and macrophages/microglia can be transcribed into cDNA for use in quantitative PCR analysis. mRNA expression levels of pcna against β-actin house-keeping gene in isolated neurons and macrophages/microglia determined by qPCR (N = 3). Fold change was measured using the comparative (ΔΔCT) method. Error bar represent mean ± SEM. Please click here to view a larger version of this figure.
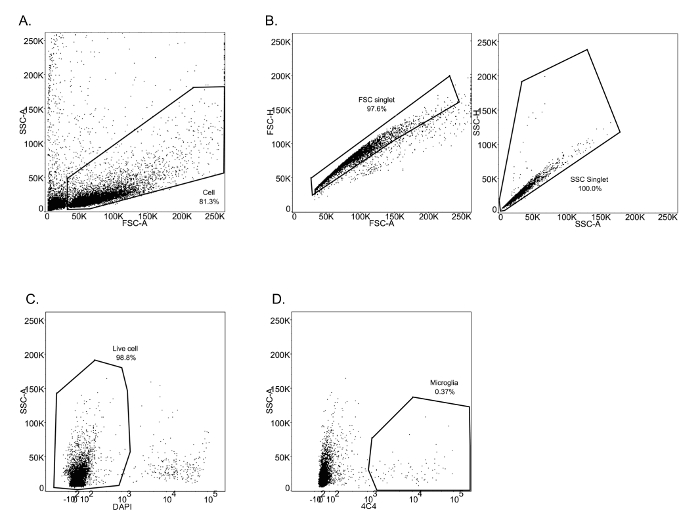
Figure 3: FACS sorting for microglia from 3 dpf zebrafish larvae. (A-C) Successive gating show sequential selection of all brain cells (A), single cells by forward scatter and side scatter (B). (C) Dead cells were excluded by DAPI labelling. (D) Microglia were identified by 4C4 positive staining. Please click here to view a larger version of this figure.
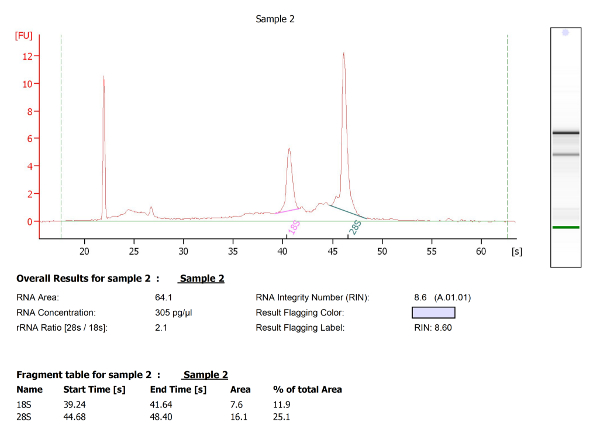
Figure 4: Micro-capillary electrophoresis results of extracted RNA from 5 dpf zebrafish microglia. The two tall peaks are the 18S and 28S ribosomal RNA. RNA integrity number (RIN) was automatically calculated by the bioanalyzer software using the generated ratio of the 18S and 28S ribosomal subunits and the analysis of the entire electrophoretic trace. Microglia RNA has a RIN 8.6. Please click here to view a larger version of this figure.
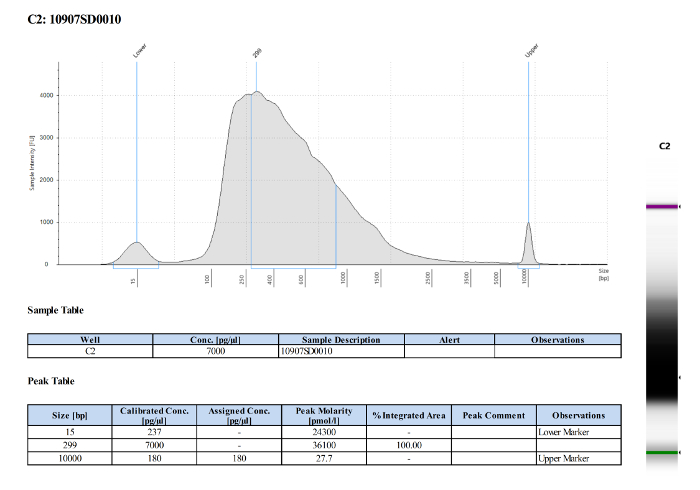
Figure 5: Quality and quantity tests of amplified cDNA from RNA sample of 5 dpf zebrafish microglia. The image shows the cDNA fragment size distribution of the analyzed sample with a mean size of 299 bp. Please click here to view a larger version of this figure.
| Condition | Experiment | Fish number | Cell number | Cell number per fish | RNA concentration (pg/ul) | Total RNA (pg) | RNA amount per cell (pg) | RIN score |
| 3dpf | 1 | 700 | 11922 | 17.03 | 126 | 1512 | 0.13 | 8 |
| 3dpf | 2 | 600 | 22527 | 37.55 | 253 | 3036 | 0.13 | 7.9 |
| 3dpf | 3 | 600 | 18688 | 31.15 | 255 | 3060 | 0.16 | 7.7 |
| 3dpf | 4 | 600 | 11121 | 18.54 | 189 | 2268 | 0.20 | 7.8 |
| 3dpf | 5 | 600 | 15581 | 25.97 | 131 | 1572 | 0.10 | 8.4 |
| 3dpf | 6 | 600 | 11965 | 19.94 | 256 | 3072 | 0.26 | 8.2 |
| 5dpf | 1 | 600 | 58629 | 97.72 | 362 | 4344 | 0.07 | 7.4 |
| 5dpf | 2 | 600 | 32510 | 54.18 | 348 | 4176 | 0.13 | 8.1 |
| 5dpf | 3 | 600 | 77884 | 129.81 | 594 | 7128 | 0.09 | 8.3 |
| 5dpf | 4 | 600 | 50755 | 84.59 | 305 | 3660 | 0.07 | 8.6 |
| 5dpf | 5 | 600 | 44967 | 74.95 | 134 | 1608 | 0.04 | 7.6 |
| 5dpf | 6 | 600 | 51031 | 85.05 | 163 | 1956 | 0.04 | 7.9 |
| 7dpf | 1 | 600 | 60496 | 100.83 | 183 | 2196 | 0.04 | 7.6 |
| 7dpf | 2 | 450 | 55517 | 123.37 | 183 | 2196 | 0.04 | 7.8 |
| 7dpf | 3 | 600 | 88897 | 148.16 | 465 | 5580 | 0.06 | 8.1 |
| 7dpf | 4 | 600 | 63008 | 105.01 | 356 | 4272 | 0.07 | 8.4 |
| 7dpf | 5 | 350 | 34956 | 99.87 | 245 | 2940 | 0.08 | 8.1 |
| 7dpf | 6 | 600 | 63887 | 106.48 | 341 | 4092 | 0.06 | 7.8 |
Table 1: Summary of microglia isolation and RNA extraction data from 3, 5 and 7 dpf zebrafish larvae.
| Internal Sample ID | External Sample ID | Qubit (ng/ul) | Qubit(ng/ul) | Average concentration (ng/ul) | Volume (ul) | ug received | Pass/fail for minimum concentration | Pass/fail for minimim quantity | Pass/fail for recommended quantity |
| 10907SD0010 | 5 dpf (experiment 4) | 58.8 | 58.4 | 58.6 | 30 | 1.76 | Pass | Pass | Pass |
| Sample Requirements for Library Prep: | |||||||||
| Library Prep | Minimum Quantity (ng) | Recommended Quantity (ng) | Minimum Concentration ng/uL | ||||||
| TruSeq Nano gel free 350 bp insert library from cDNA | 600 | 1100 | 10 | ||||||
Table 2: Quantity tests of amplified cDNA from RNA sample of 5 dpf zebrafish microglia.
