The Use of Mouse Splenocytes to Assess Pathogen-associated Molecular Pattern Influence on Clock Gene Expression
Summary
This protocol describes a technique using mouse splenocytes to discover pathogen-associated molecular patterns that alter molecular clock gene expression.
Abstract
From behavior to gene expression, circadian rhythms regulate nearly all aspects of physiology. Here, we present a methodology to challenge mouse splenocytes with the pathogen-associated molecular patterns (PAMPs) lipopolysaccharide (LPS), ODN1826, and heat-killed Listeria monocytogenes and examine their effect on the molecular circadian clock. Previously, studies have focused on examining the influence of LPS on the molecular clock using a variety of in vivo and ex vivo approaches from an assortment of models (e.g., mouse, rat, and human). This protocol describes the isolation and challenge of splenocytes, as well as the methodology to assess clock gene expression post-challenge via quantitative PCR. This approach can be used to assess not only the influence of microbial components on the molecular clock but other molecules as well that may alter expression of the clock. This approach could be utilized to tease apart the molecular mechanism of how PAMP-Toll-like receptor interaction influences clock expression.
Introduction
The master clock in mammals, which orchestrates 24 h oscillations for nearly all aspects of physiology and behavior, is located within the suprachiasmatic nucleus (SCN) of the hypothalamus1,2. In addition to regulating biological processes on an organismal level, the master clock also synchronizes peripheral cellular clocks throughout the body3,4,5. While the molecular clock machinery consists of at least three interlocking transcriptional-translational feedback loops, the core is comprised of the Period (Per1-3), Cryptochrome (Cry1-2), Bmal1, and Clock genes6,7. Besides maintaining the accurate timing of the core molecular clock, some ancillary clock gene products (e.g., Rev-erbα and Dbp) also regulate expression of non-clock genes, i.e., clock controlled genes (CCGs)6,7.
Functional molecular clocks have been described in various immune tissues (e.g., spleen and lymph nodes)8 and cells (e.g., B cells, dendritic cells, macrophages)8,9. These cells detect and respond to pathogen-associated molecular patterns (PAMPs), conserved microbial components, via innate immune recognition receptors such as Toll-like receptors (TLRs)10. To date, 13 functional TLRs have been described, which recognize microbial constituents such as bacterial cell wall components, flagellar protein, and microbial nucleic acids10. The PAMP, lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria recognized by TLR4, has been shown to alter circadian rhythms at the both organismal and molecular levels. For example, in vivo challenge of LPS induced photic-like phase delays as measured by activity in mice11 and led to reduced clock gene expression in the SCN and liver as determined by in situ hybridization and quantitative PCR, respectively, in rats12. After an in vivo challenge with LPS, analysis of human peripheral blood leukocytes13 and subcutaneous adipose tissue14 revealed altered expression of several clock genes as measured via qPCR. Lastly, ex vivo LPS challenges of human macrophages and mouse peritoneal macrophages, also led to altered clock expression as measured by qPCR14.
Here, we describe a protocol to assess the influence of the PAMPs LPS, ODN1826 (synthetic oligonucleotides containing unmethylated CpG motifs), and heat-killed Listeria monocytogenes (HKLM), recognized by TLR4, TLR9, and TLR2, respectively, on molecular clock gene expression in mouse splenocytes. The protocol includes mouse splenectomy, splenocyte isolation and challenge, RNA extraction, cDNA synthesis, and qPCR to assess expression of several clock genes. This protocol allows for the timely acquisition of a large number of immune cells with very little animal or cellular manipulation, which can then be challenged ex vivo with various PAMPs. The molecular clock has been shown to modulate various aspects of the immune response8,15,16, therefore, disruption of the molecular clock would most likely impair the proper time-dependent variation of the immune response. In addition, since disruptions of circadian rhythms can lead to serious pathologies17,18,19,20, it may be of interest for researchers to challenge splenocytes with a wide range of molecules and assess their influence on the clock.
Protocol
During the study, animal care and treatment complied with National Institutes of Health policy, were in accordance with institutional guidelines, and were approved by the University of Hartford Animal Institutional Animal Care and Use Committee.
1. Entrainment of Animals
NOTE: Twenty week-old male B6129SF2/J mice are used in the study.
- Entrain mice to a 12 h light (standard overhead white light)/12 h dark cycle for 2 weeks prior to the experiment.
NOTE: Here zeitgeber time (ZT) 0 corresponds to lights on and ZT12 to lights off, while keeping all other environmental factors (i.e., food, water, and room temperature) constant 21.
2. Preparation of Instruments, Culture Medium, and Challenge Medium
- Autoclave forceps and dissecting scissors. Wrap pairs of frosted microscope slides in aluminum foil and autoclave.
- Prepare culture medium by adding fetal bovine serum (FBS) to RPMI 1640 to a final concentration of 10%. Prepare 10 mL of challenge medium in 50 mL tubes by adding the following PAMPs to the culture medium (RPMI with 10% FBS); LPS (5 µg/mL), ODN1826 (5 µg/mL), heat-killed Listeria monocytogenes (108 HKLM/mL), or another PAMP in its suggested concentration.
- Warm culture medium and challenge medium to 37 °C in a water bath, and keep approximately 70 mL of sterile phosphate buffered saline (PBS, pH 7.2) at room temperature.
- Add 10 mL of culture medium using a 10 mL pipette and pipette aid to a 50 mL tube and place on ice.
- Add approximately 30 mL of 70% ethanol to a 100 mL beaker and place ends of dissecting scissors and forceps into the beaker to prevent microbial contamination.
- Prepare lysis buffer for RNA isolation by adding 10 µL β-Mercaptoethanol (under a fume hood) to every 1 mL of Buffer RLT in a 50 mL tube. Make only the amount that will be needed (600 µL per sample, which consists of approximately 1 x 106 cells).
NOTE: The Buffer RLT is a component of the RNA extraction kit that supports the binding of RNA to the silica membrane.
3. Splenocyte Isolation and Challenge
- Euthanize mice at a particular zeitgeber time via narcosis by keeping them in their original cage and adding CO2 to the cage at a flow rate of 3 L/min. Continue supplying CO2 for 1 min after breathing stops.
- Confirm death via cervical dislocation by placing the thumb and index finger on either side of the neck at the base of the skull. Alternatively, press a rod at the base of the skull while quickly pulling (using the other hand) the base of the tail or the hind limbs to cause separation of the cervical vertebrae from the skull22.
- Spray the mouse trunk with 70% ethanol and wipe with a paper towel. Place the mouse on its back and slightly tilted onto its right side. Cut away the fur, using dissecting scissors, along the mouse's left side, about halfway between the front and back legs.
- Using forceps, grab the peritoneum and carefully make an incision so as not to damage the spleen. Remove spleen with forceps and place into a sterile 50 mL tube containing approximately 10 mL of culture media on ice. Repeat for the remaining animals.
NOTE: The spleen is the color of a kidney bean, and it is longer and flatter than the kidney. - Transfer one spleen with 2 mL of culture medium to a small sterile Petri dish.
- Homogenize the spleen by grinding it between the frosted portion of two sterile frosted slides. If possible, keep the issue and cells in the medium during the homogenization process.
- Once thoroughly homogenized, pipet the 2 mL of culture medium containing the splenocytes through a 40 µm nylon cell strainer into a 50 mL tube.
- Repeat the above steps for the remaining spleens using new pairs of frosted slides, 50 mL tubes with culture medium, and cell strainers.
- Add 8 mL of cold culture medium to each of the 50 mL tubes containing the splenocytes for a total volume of 10 mL/tube. Determine the number of cells per milliliter using a hemocytometer.
- Add approximately 1 x 106 cells/well to 6-well culture plates. Add cells to the number of wells that correspond to the number of different PAMPs being used for the experiment and include a control well. Add 3 mL of culture medium or 3 mL of challenge medium to the respective wells.
- Incubate the plates at 37 °C in 5% CO2 for 3 h.
- Scrape the cells from the bottom of the well using a 1,000 µL pipet tip attached to a P1000 micropipette. Transfer the medium containing the cells using the same 1,000 µL pipet tip to a 15 mL tube.
- Pellet the cells via centrifugation at 167 x g for 5 min at room temperature. Remove the supernatant and wash the cell pellet with 5 mL of PBS.
- Pellet the cells a second time at 167 x g for 5 min, remove the supernatant, and add 600 µL of lysis buffer to the cell pellet in order to lyse the cells. Then proceed to RNA isolation.
4. RNA Isolation and cDNA Synthesis
- Isolate RNA from splenocytes using the RNA extraction kit according to manufacturer's instructions and perform the 'optional' on-column DNA digestion.
- Prior to cDNA synthesis, determine RNA concentration using a microvolume spectrophotometer to verify that the concentration is within the optimal RNA range (up to 2 µg) of the cDNA synthesis kit.
- Synthesize cDNA for each of the samples using the reverse transcription kit according to manufacturer's instructions. Use 10 µL of RNA for each of the samples in a 20 µL total reaction volume. At the completion of the reverse transcription (thermocycler run) add 20 μL H2O to each reaction.
- Construct the standard curving using a P10 or P20 micropipette to pool mRNA from a few control samples (e.g., 5 µL from 2 samples for a total of 10 µL) into a 0.5 mL tube to prepare the cDNA.
- Add 10 µL 2x reverse transcriptase master mix.
- At the completion of the reverse transcription, add 10 µL of H2O to the reaction tube, which will serve as the starting concentration ("1") in the dilution series for the standard curve.
- Perform a 10-fold dilution series (1 to 10-4) by adding 45 µL of water using a P100 or P200 micropipette into four 0.5 mL tubes designated 10-1, 10-2, 10-3, and 10-4.
- Add 5 µL of the starting concentration "1" into the first tube (10-1) using a P20 micropipettor, mix by pipetting up and down several times, then transfer 5 µL from the 10-1 tube into the tube designated 10-2 and mix.
- Transfer 5 µL using a P20 micropipette from the 10-2 tube into the tube designated 10-3 and mix. Transfer 5 µL using a P20 micropipettor from the 10-3 tube into the tube designated 10-4 and mix.
5. Quantitative Polymerase Chain Reaction (qPCR)
- Determine relative quantitation of mRNA levels by qPCR. Within the experimental set up, select "Quantitation – Relative Standard Curve" and "TaqMan" chemistry. Change the reaction volume to 10 µL. Enter the relevant information into the plate layout (e.g., target gene, standard curve, reporter, etc.).
- Prepare the reaction so that it contains 0.5 µL of the primer/probe assay, 5 µL of gene expression master mix, 2 µL of H2O and 2.5 µL of cDNA (10–100 ng).
- Prepare a master mix by multiplying each of the preceding reagents (except the cDNA) by the number of reactions, making sure to include those from the standard curve, negative controls, performing each reaction in duplicate, and 2 extra reactions that will account for pipetting variation.
- Pipet 7.5 µL of the prepared master mix into pre-determined wells in a 96-well reaction plate with a micropipette or a multichannel pipette. Pipet 2.5 µL of cDNA into the appropriate well for the unknowns and standards, and 2.5 µL of H2O into the negative control wells.
- Include primer/probe assays for various molecular clock genes, and also include an assay for an endogenous control (e.g., β-actin).
- In order to determine relative expression values, calculate the mean relative quantity for each replicate, then, for each sample, divide the mean relative quantity for the target gene by the mean relative quantity for β-actin.
6. Statistical Analysis
- Using statistical analysis software, enter data into the program under the column statistics option. Select a one-way ANOVA with the Dunnett's post hoc test to assess differences between mean values of clock gene expression after PAMP challenge versus the control.
Representative Results
Mice were sacrificed at ZT13, splenocytes were isolated and challenged ex vivo with the PAMPs LPS, ODN1826, or HKLM. After 3 h, RNA was isolated, and qPCR was used to assess relative expression levels of the molecular clock genes Clock, Per2, Dbp, and Rev-erbα compared to unchallenged control cells. After PAMP challenge, Clock expression levels were not significantly different than expression in the control cells (Figure 1A). Per2 expression levels were significantly elevated in cells challenged with LPS and ODN1826 when compared to unchallenged controls (Figure 1B). LPS was the only PAMP to alter Rev-erbα expression, as mRNA levels were significantly lower than in the unchallenged controls (Figure 1C). Lastly, significantly lower mRNA levels were observed for Dbp after challenge with each of the PAMPs when compared to the controls (Figure 1D). Consistent with what has been previously shown, out of all the clock associated genes examined, Dbp expression tends to be most affected by PAMP challenge23.
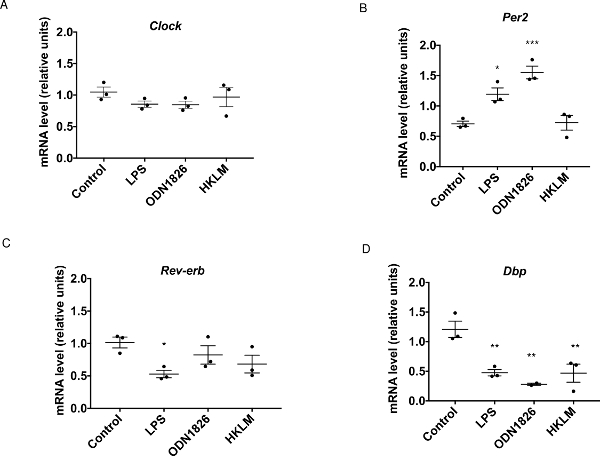
Figure 1: Altered clock gene expression in mouse splenocytes after ex vivo PAMP challenge. Splenocytes were isolated at ZT13 and challenged with LPS, ODN1826, or HKLM. Relative clock mRNA levels (normalized to β-actin) were determined by qPCR 3 h after challenge. Each data point represents expression level for 1 animal. Experimental mean + SEM are given. *p <0.05, **p <0.01, ***p <0.001. The indicated challenges were significantly different from the control (unchallenged cells) as per one-way ANOVA with the Dunnett's post hoc test. Please click here to view a larger version of this figure.
Discussion
Within this protocol, a microvolume spectrophotometer can be used to quantify and assess the purity of the RNA being used in determining gene expression. Nucleic acids absorb UV light at 260 nm, proteins typically absorb light at 280 nm, while other potential contaminants used during an RNA extraction procedure (e.g., phenol) are detectable at 230 nm. Therefore, by assessing the absorbance (A) ratio at 260/280 nm (RNA to protein) and 260/230 nm (RNA to non-protein contaminants) the quality of the RNA can be assessed. High quality RNA has an A260/280 ratio between 1.8–2.1, as lower ratios indicate protein contamination. A pure RNA sample will have an A260/230 ratio of 2.0.
When determining the relative expression of a target gene (i.e., Per2, Clock, Rev-erbα, and Dbp), an endogenous control gene (a gene in which expression levels do not differ between samples) must also be selected. Relative expression of the target gene is then normalized to the expression of the endogenous control gene. Differences in starting material (number of splenocytes), variation in reverse-transcriptase efficiency, varying rates of RNA degradation, etc., will be corrected for by the endogenous control gene (β-actin in this protocol). However, it is wise to verify that the treatment being examined does not alter expression of the endogenous control gene. This can be accomplished by assessing β-actin levels from several replicates of an equal number of cells (treatment vs. non-treatment). In theory, their β-actin levels should be identical. Another approach to guard against endogenous control variation would be to use a panel of endogenous controls (e.g., β-actin, Gapdh, and 18S rRNA gene).
When examining the impact of PAMPs on the molecular clock, the time of day when mice are euthanized and splenocytes are subsequently challenged must be taken into consideration. Tlr expression and responsiveness has previously been shown to demonstrate time-of-day dependent variation8,15, therefore, a time of day when TLR responsiveness is at its peak, could result in a greater influence on the clock. Furthermore, expression of molecular clock genes will also fluctuate throughout the day in splenocytes, therefore, a reduction of clock gene expression due to PAMP challenge would be most significant if examined during the time of peak expression9. Since Dbp and Rev-erbα have been shown to demonstrate significant expression peaks in splenocytes and splenic immune cells around the light-dark interphase 8,9,23,24 (ZT12), in the current method, cells were isolated and challenged at ZT13 in order to have a greater chance at detecting a reduction in these genes. Conversely, a PAMP that could increase clock expression, would most likely be observed if looking at a time of day when clock expression is at its lowest.
Since mice are nocturnal animals, their rest phase is during the light period, which corresponds to human activity. Therefore, ideally, mice will be housed in a room with minimal traffic and one that contains a white-noise machine in order to reduce daytime disturbances as this could disrupt the circadian rhythms of the mice. Furthermore, cage changes should be done well in advance of the experiment date. Any work in the animal room (including euthanasia of the animals) during the dark period, should be conducted under red light in order to avoid disrupting the rhythms of the mice.
Diurnal rhythms are subjected to environmental stimuli (e.g., light or food), which are termed zeitgebers. In the case of a 12 h light/12 h dark cycle, the zeitgeber (i.e., light) resets the clock to a 24 h period. While most diurnal rhythms are circadian (i.e., daily rhythms that occur in the absence of an external cue), they are not true circadian rhythms until they have been shown to oscillate with an approximate 24 h period under constant environmental conditions. Therefore, this procedure could be performed using mice under constant conditions, which would entail entraining mice to the light-dark cycle as described above, but then holding the animals in constant darkness for 3 days prior to sampling. This type of experiment is referred to as a dark-dark (DD) experiment and the time point of sampling would be referred to as CT (circadian time), not ZT.
While this method can identify PAMPs that alter clock gene expression within splenocytes, it does not take into account how these PAMPs affect the master clock or peripheral clocks throughout the body. Since the spleen is composed of a heterogeneous population of cells, individual PAMPs could impact each cell type differently. For example, Tlr9 expression rhythms in the mouse spleen differ between splenocytes, macrophages, B cells, and DCs15. Additionally, Tlr1, Tlr3, Tlr4, Tlr6, Tlr7,and Tlr8 displayed significant daily oscillations in an adherent splenocyte population but only Tlr2 and Tlr6 experience daily oscillations in enriched splenic macrophages24. Therefore, in order to investigate the outcome of a challenge on individual cell types, cells could be isolated via magnetic cell sorting, as previously described9,15 and then subsequently challenged. Additionally, the splenic cell population fluctuates over the daily cycle, which could also play a role in sensitivity to a particular PAMP and subsequent impact on the clock8.
This method allows for the isolation of a large number of immune cells that consist predominately of B cells (~58%), T cells (~21%), dendritic cells (~5%), and macrophages (~4%)25. The large number of cells provides the opportunity to challenge splenocytes with a variety of PAMPs in a single experiment. The splenocyte isolation procedure is very easy to perform, can be completed within minutes (depending on the number of animals), and with minimal animal or cellular manipulation, which is essential when examining the molecular clock because as mentioned above, these actions can disrupt the timing of the clock as well as clock-controlled genes. The results for this procedure were highly reproducible, as significance between challenged and unchallenged cells was achieved with just three animals and the results were consistent with previously published work23 (Figure 1). It should be noted that increasing the number of animals per group might have revealed statistically significant differences between a challenge group and control (e.g., ODN 1826 and Rev-erbα).
Moving forward, while this protocol only addresses the acute effects on clock gene expression after PAMP challenge, it could provide proof of principle for further investigation. For example, this assay could be used as a model to decipher the molecular mechanisms regarding TLR – PAMP interaction and how it influences the molecular clock. It could also be used to determine the length of time it takes for the molecular clock to recover after a PAMP challenge, which could be determined by conducting a time-course experiment (i.e., assessing expression after varying times post challenge). As mentioned above, subsequent experiments could be performed to examine PAMP challenge on specific splenocyte cell populations. Since several pathogens stimulate multiple TLRs upon infection, it would be interesting to use this protocol to investigate if challenging with multiple PAMPs have a synergistic effect on clock gene expression.
Divulgations
The authors have nothing to disclose.
Acknowledgements
This work was supported by Faculty Research grants from the College of Arts and Sciences Dean’s Office at the University of Hartford.
Materials
| Frosted slides | Fisher | 12-550-343 | |
| Cell strainers | Fisher | 22363547 | |
| Lipopolysaccharide | InvivoGen | ltrl-eklps | |
| ODN1826 | InvivoGen | Tlrl-1826-1 | |
| HKLM | InvivoGen | Tlrl-hklm | |
| RPMI 1640 | Gibco | 11875-093 | |
| PBS | Gibco | 20012-043 | |
| RNeasy Mini Kit | Qiagen | 74104 or 74106 | |
| RNase-Free DNase Set | Qiagen | 79254 | |
| 6-well cell culture plate | Denville | T1006 | |
| 50 ml tubes | Corning | 352070 | |
| 15 ml tubes | Corning | 352097 | |
| High Capacity cDNA Reverse Transcription Kit | ThermoFisher | 4368814 | |
| TaqMan Gene Expression Assays b-actin | ThermoFisher | Mm00607939_s1 | |
| TaqMan Gene Expression Assays Per2 | ThermoFisher | Mm00478113_m1 | |
| TaqMan Gene Expression Assays Rev-erba | ThermoFisher | Mm00520708_m1 | |
| TaqMan Gene Expression Assays Bmal1 | ThermoFisher | Mm00500226_m1 | |
| TaqMan Gene Expression Assays Dbp | ThermoFisher | Mm00497539_m1 | |
| qPCR machine StepOnePlus | ThermoFisher | ||
| TaqMan Gene Expression Master Mix | ThermoFisher | 4369016 | |
| MicroAmp Fast 96-well reaction plate (0.1 ml) | ThermoFisher | 4346907 | |
| Statistical Analysis Software | Prism 7.0a |
References
- Bell-Pedersen, D., et al. Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat. Rev. Genet. 6, 544-556 (2005).
- Mohawk, J. A., Green, C. B., Takahashi, J. S. Central and Peripheral Circadian Clocks in Mammals. Annu. Rev. Neurosci. 35, 445-462 (2012).
- Balsalobre, A., Damiola, F., Schibler, U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 93, 929-937 (1998).
- Yoo, S. -. H., et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. U. S. A. 101, 5339-5346 (2004).
- Yamazaki, S. Resetting Central and Peripheral Circadian Oscillators in Transgenic Rats. Science. 288, 682-685 (2000).
- Lowrey, P. L., Takahashi, J. S. Genetics of circadian rhythms in mammalian model organisms. Adv. Genet. 74, 175-230 (2011).
- Curtis, A. M., Bellet, M. M., Sassone-Corsi, P., O’Neill, L. A. J. Circadian Clock Proteins and Immunity. Immunity. 40, 178-186 (2014).
- Keller, M., et al. A circadian clock in macrophages controls inflammatory immune responses. Proc. Natl. Acad. Sci. U. S. A. 106, 21407-21412 (2009).
- Silver, A. C., Arjona, A., Hughes, M. E., Nitabach, M. N., Fikrig, E. Circadian expression of clock genes in mouse macrophages, dendritic cells, and B cells. Brain. Behav. Immun. 26, 407-413 (2012).
- Kawai, T., Akira, S. The role of pattern-recognition receptors in innate immunity update on Toll-like receptors. Nat. Publ. Gr. 11, 373-384 (2010).
- Marpegán, L., Bekinschtein, T. A., Costas, M. A., Golombek, D. A. Circadian responses to endotoxin treatment in mice. J. Neuroimmunol. 160, 102-109 (2005).
- Okada, K., et al. Injection of LPS Causes Transient Suppression of Biological Clock Genes in Rats. J. Surg. Res. 145, 5-12 (2008).
- Haimovich, B., et al. In vivo endotoxin synchronizes and suppresses clock gene expression in human peripheral blood leukocytes. Crit. Care Med. 38, 751-758 (2010).
- Curtis, A. M., et al. Circadian control of innate immunity in macrophages by miR-155 targeting Bmal1. Proc. Natl. Acad. Sci. U. S. A. 112, 7231-7236 (2015).
- Silver, A. C., Arjona, A., Walker, W. E., Fikrig, E. The Circadian Clock Controls Toll-like Receptor 9-Mediated Innate and Adaptive Immunity. Immunity. , (2012).
- Gibbs, J. E., et al. The nuclear receptor REV-ERB α mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. PNAS. 109, 582-587 (2012).
- Zee, P. C., Attarian, H., Videnovic, A. Circadian rhythm abnormalities. Contin. Lifelong Learn. Neurol. 19, 132-147 (2013).
- Bovbjerg, D. H. Circadian disruption and cancer: sleep and immune regulation. Brain. Behav. Immun. 17, 48-50 (2003).
- Fu, L., Lee, C. C. The circadian clock: pacemaker and tumour suppressor. Nat. Rev. Cancer. 3, 350-361 (2003).
- Germain, A., Kupfer, D. J. CIRCADIAN RHYTHM DISTURBANCES IN DEPRESSION. Hum. Psychopharmacol. 23, 571-585 (2008).
- . Basic Mouse Care and Maintenance. JoVE. , (2018).
- Leary, S., et al. AVMA Guidelines for the Euthanasia of Animals : 2013 Edition. AVMA. , (2013).
- Silver, A. C. Pathogen-associated molecular patterns alter molecular clock gene expression in mouse splenocytes. PLoS One. , 12-15 (2017).
- Silver, A. C., et al. Daily oscillations in expression and responsiveness of Toll-like receptors in splenic immune cells. Heliyon. , 00579 (2018).
