Synthesis and Structure Determination of µ-Conotoxin PIIIA Isomers with Different Disulfide Connectivities
Summary
Cysteine-rich peptides fold into distinct three-dimensional structures depending on their disulfide connectivity. Targeted synthesis of individual disulfide isomers is required when buffer oxidation does not lead to the desired disulfide connectivity. The protocol deals with the selective synthesis of 3-disulfide-bonded peptides and their structural analysis using NMR and MS/MS studies.
Abstract
Peptides with a high number of cysteines are usually influenced regarding the three-dimensional structure by their disulfide connectivity. It is thus highly important to avoid undesired disulfide bond formation during peptide synthesis, because it may result in a completely different peptide structure, and consequently altered bioactivity. However, the correct formation of multiple disulfide bonds in a peptide is difficult to obtain by using standard self-folding methods such as conventional buffer oxidation protocols, because several disulfide connectivities can be formed. This protocol represents an advanced strategy required for the targeted synthesis of multiple disulfide-bridged peptides which cannot be synthesized via buffer oxidation in high quality and quantity. The study demonstrates the application of a distinct protecting group strategy for the synthesis of all possible 3-disulfide-bonded peptide isomers of µ-conotoxin PIIIA in a targeted way. The peptides are prepared by Fmoc-based solid phase peptide synthesis using a protecting group strategy for defined disulfide bond formation. The respective pairs of cysteines are protected with trityl (Trt), acetamidomethyl (Acm), and tert-butyl (tBu) protecting groups to make sure that during every oxidation step only the required cysteines are deprotected and linked. In addition to the targeted synthesis, a combination of several analytical methods is used to clarify the correct folding and generation of the desired peptide structures. The comparison of the different 3-disulfide-bonded isomers indicates the importance of accurate determination and knowledge of the disulfide connectivity for the calculation of the three-dimensional structure and for interpretation of the biological activity of the peptide isomers. The analytical characterization includes the exact disulfide bond elucidation via tandem mass spectrometry (MS/MS) analysis which is performed with partially reduced and alkylated derivatives of the intact peptide isomer produced by an adapted protocol. Furthermore, the peptide structures are determined using 2D nuclear magnetic resonance (NMR) experiments and the knowledge obtained from MS/MS analysis.
Introduction
The use of bioactive peptides in pharmaceutical research and development is highly recognized, because they represent potent and highly selective compounds for specific biological targets1. For their bioactivity, however, the three-dimensional structure is of great importance in order to perform structure-activity relationship studies2,3,4. Apart from the primary amino acid sequence that influences the overall conformation, disulfide bonds significantly stabilize the structure of cysteine-rich peptides5. Multiple disulfide-bridged peptides include conotoxins such as µ-PIIIA from Conus purpurascens which contains six cysteines in its sequence. This high cysteine content theoretically allows the formation of 15 disulfide isomers. The correct disulfide connectivity is very important for the biological activity6,7. However, the question that arises is whether there is more than one bioactive conformation of naturally occurring peptides and if so, which of those isomers possesses the highest biological activity? In the case of µ-conotoxins, the biological targets are voltage-gated sodium ion channels, and µ-PIIIA in particular is most potent for subtypes NaV1.2, NaV1.4 and NaV1.73.
The synthesis of disulfide-bridged peptides can be achieved using various methods. The most convenient method for the formation of disulfide bonds within a peptide is the so-called oxidative self-folding approach. Here, the linear precursor of the desired cyclic peptide is synthesized first using solid-phase peptide synthesis, which is after the cleavage from the polymeric support subjected to oxidation in a buffer system. Redox-active agents such as reduced and oxidized glutathione (GSH/GSSG) are often added to promote the formation of the disulfide bonds. The main disadvantage of buffer-supported self-folding is that the disulfide bonds are not formed selectively in a stepwise fashion. Compared to the native peptide, for which often only one specific disulfide isomer is described, it is possible to obtain numerous other isomers with this approach8. µ-PIIIA has already been shown to result in at least three differently folded isomers upon self-folding in a previous study3. The separation of such an isomer mixture is rather difficult due to similar retention times if using chromatographic purification methods9. The targeted synthesis of a specific isomer is therefore advantageous. To specifically produce an isomer with defined disulfide connectivity, a special strategy is required in which the disulfide bonds are successively closed. Therefore, the linear precursor carrying distinct protecting groups at the individual cysteine pairs is synthesized on the polymer support. After the elimination, the cysteine pairs are individually and successively deprotected and linked in an oxidation reaction to yield the desired disulfide bonds10,11,12,13,14,15,16. After the synthesis and the purification of the reaction product, it is required to confirm the identity and the disulfide connectivity by suitable analytical methods. Numerous analytical methods are available for the elucidation of the primary amino acid sequence, e.g., MS/MS, while the determination of the disulfide connectivity still remains much less investigated. Apart from the complexity of such multiple disulfide-bonded peptides, product-related impurities (e.g., from disulfide scrambling), due to sample preparation and work-up can further complicate analysis. In this paper, we show that the use of a combination of different analytical techniques is necessary to unequivocally clarify the identity of the disulfide bonds in the µ-PIIIA isomers. We have combined chromatographic methods with mass spectrometry and provided the same samples to NMR spectroscopy. In matrix-assisted laser desorption/ionization(MALDI) MS/MS analysis, we identified the disulfide bonds by using partial reduction and iodoacetamide derivatization because top-down analysis is not possible for this peptide. 2D NMR experiments were performed in order to obtain a three-dimensional structure of each isomer. Thus, by combining distinct sophisticated analytical methods, it is possible to elucidate properly the disulfide connectivity and three-dimensional structure of complex multiple disulfide-bonded peptides7.
Protocol
Note: All amino acids used herein were in the L-configuration. The abbreviations of amino acids and amino acid derivatives were used according to the recommendations of the Nomenclature Committee of IUB and the IUPAC-IUB Joint Commission on Biochemical Nomenclature.
1. Solid-Phase Peptide Synthesis (SPPS)
NOTE: Carry out the synthesis with a solid-phase peptide synthesizer. Perform the synthesis of the linear peptide precursors of the general sequence ZRLCCGFOKSCRSRQCKOHRCC-NH2 using a standard protocol for 9-fluorenylmethyloxycarbonyl (Fmoc) chemistry. Apply the following protected amino acids: Pyr(Boc (tert-butyloxycarbonyl)), Arg(Pbf (2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl)), Asn(Trt), Asp(tBu), Hyp(tBu), Lys(Boc), Ser(tBu), Gln(Trt), Glu(tBu), Trp(Boc), Tyr(tBu), Thr(tBu), and His(Trt). Protect the cysteine pairs with Trt-, Acm-, or tBu-groups according to the intended disulfide connectivity.
- Preparation
- Dry the Fmoc Rink-amide resin (loading: 0.28 mmol/g) using a lyophilizer overnight.
- Enter the desired peptide sequence (1-letter code) into the program of the synthesizer. The program calculates the required amount for every individual reagent and indicates the solvent quantities.
- Weigh the individual reagents (amino acids, HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate)) according to the protocol and dissolve them in dimethylformamide (DMF) to a final concentration of 0.6 M (amino acids) and 0.6 M (HBTU), respectively.
- Transfer the different reagents (amino acids, HBTU, N-methylmorpholine (NMM, 50% in DMF), piperidine (20% in DMF), DMF, dichloromethane (DCM)) to the corresponding vessels and place them in the appropriate rackets of the solid-phase peptide synthesizer.
- Add 100 mg of dry resin to the reaction columns and put them in the racket of the synthesizer. Start the solid-phase peptide synthesis.
- SPPS-Protocol (provided by synthesizer)
NOTE: The standardized protocol usually applies to 100 mg of resin (loading: 0.53 mmol/g) added to one reaction column for 53 μmol scale leading to the following equivalents: 5 eq. HBTU, 10 eq. NMM, 5 eq. amino acid. In case of PIIIA, however, a loading of 0.28 mmol/g (28 µmol scale) is used, which results in the specified higher equivalents.- Preparation of the resin
- Rinse the resin with 2500 µL of DMF, 1400 µL of DCM, and 1400 µL of DMF.
- Flush the resin with air until the solvent is removed and rinse the resin with 2500 µL of DMF.
- Cleavage of the Fmoc protecting group
- Add 20% piperidine in DMF (1000 µL) to the resin and wait for 6 min. Remove the solution from the resin. Repeat Step 1.2.2.1.
- Rinse twice with DMF (1st 4000 µL, 2nd1400 µL), flush the resin with air until the solvent is removed and rinse twice with 2000 µL of DMF.
- Coupling reaction
- Mix the following reagents in a separate vial: HBTU (415 µL; 0.6 M in DMF; 249 µmol; 9 eq.), NMM (112 µL; 50% in DMF; 510 µmol; 18 eq.), Fmoc-amino acid (420 µL; 0.6 M in DMF; 252 µmol; 9 eq.). Add the mixture to the resin and wait for 13 min. Remove the solution from the resin. Repeat Step 1.2.3.1.
- Wash with 3000 µL of DMF. Wash twice with 1400 µL of DMF. Wash twice with 2000 µL of DMF.
- Repeat Steps 1.2.2 and 1.2.3 according to the number of amino acids in the peptide sequence.
- Final Fmoc cleavage and resin wash
- Add 20% piperidine in DMF (1000 µL) to the resin and wait for 6 min. Remove the solution from the resin. Repeat Step 1.2.4.1.
- Rinse twice with DMF (1st 4000 µL, 2nd 1400 µL) and flush the resin with air. Rinse twice with 2000 µL of DMF, 4 times with 1400 µL of DCM, and flush twice with air.
- Preparation of the resin
- Work-up
- Lyophilize the resin from the reaction columns overnight after the synthesis is complete.
2. Peptide Cleavage from the Resin (Figure 1A)
NOTE: During the cleavage procedure, all amino acid side chains except for Cys(Acm) and Cys(tBu) will be deprotected. The protocol applies to 100 mg of resin.
- Combine the freeze-dried resin in a 12 ml tube and cool it to 0 °C on ice.
- Add 150 µL of a scavenger mixture (prepared from 0.75 g phenol, 0.5 mL thioanisole, 0.25 mL ethanedithiol) and 1 mL of trifluoroacetic acid (TFA, 95% in water (H2O)) on ice to the resin. Leave gently shaking for 3 h at room temperature.
- Filter the mixture through a glass frit and collect the filtrate in tubes individually filled with ice-cold diethyl ether (1 mL of cleavage mixture per 8-10 mL of diethyl ether). The peptide precipitates as a white solid.
- Rinse the filter cake with additional TFA (95% in H2O, approx. 1-3 mL).
- Close the tubes containing the precipitate and centrifuge (3400 x g) the suspensions for 1 min, decant the supernatant and wash the pellets with 8-10 mL of ice-cold diethyl ether. Repeat this step 3 times.
- Leave the pellets standing without stopper for 5 min to remove remaining traces of diethyl ether. Dissolve the crude product pellets in 1 mL of tert-butanol (80% in H2O). Freeze-dry the peptides (-80 °C).
3. Purification of the Linear Precursor with Semi-preparative High-performance liquid chromatography (HPLC)
NOTE: Purify the crude peptides by semi-preparative reversed phase (RP) HPLC equipped with a C18 column (5 µm particle size, 100 Å pore size, 250 x 32 mm) and a 3.6 mL injection loop. Use a gradient elution system of 0.1% TFA in H2O (eluent A) and 0.1% TFA in acetonitrile (MeCN)/H2O (9:1, eluent B). Detect the peaks at 220 nm.
- Add approx. 70 mg of the crude peptide to a 15 mL tube and dissolve the solid peptide in the volume of the HPLC sample loop (e.g., 3.6 mL). Use a mixture of 0.1% TFA in MeCN/H2O (1:1). Vortex until complete dissolution and centrifuge (3400 x g) the solution for 1 min.
- Draw up the sample (3.6 mL) in a 5 mL syringe and inject the sample without any air bubbles into the injection loop. Inject into the HPLC system. Separate the peptide mixture using a gradient of 0-50% eluent B over 120 min at a flow rate of 10 mL/min.
- Collect the fractions in individual tubes as they appear. After the run is complete, prepare selected fractions for mass spectrometry (MS) and HPLC analysis (Step 6.1-6.2). Freeze-dry the fractions and store them at -20 °C.
- After MS and HPLC analysis, combine the pure fractions of linear peptide and prepare the samples for the first oxidation.
4. Selective Formation of the Disulfide Bonds
- 1 st Oxidation (Figure 1B)
NOTE: During the peptide cleavage from the resin, the Cys(Trt) are deprotected, leading to two unprotected Cys residues which are subsequently subjected to oxidation to form the first disulfide bond. The following protocol applies to 15 mg of linear purified peptide (2864.5 g/mol; 5.2 µmol; 1 eq.).- Dissolve the linear peptide (15 mg) in 105 mL of an isopropanol/H2O-mixture (1:2; 0.05 mM; pH 8.5 adjusted with sodium hydroxide (NaOH)) and leave gently shaking in air under basic conditions for 12-48 h.
- Monitor the oxidation reaction via HPLC and MS. Confirm the formation of the first bridge by iodoacetamide (IAA) derivatization (Step 6).
- Freeze-dry the peptide and use it without further purification for the 2nd oxidation.
- 2nd Oxidation (Figure 1C)
NOTE: During the second oxidation, the deprotection of the Acm-protected cysteines and the formation of the second bridge are catalyzed by iodine. The protocol applies to 15 mg of peptide after the 1st oxidation (2862.5 g/mol; 5.2 µmol; 1 eq.)- Dissolve the peptide (15 mg; final concentration of 0.05 mM) in 105 mL of an isopropanol/H2O/1 M hydrochloric acid (HCl) mixture (80:12.5:7.5).
- Add 158 µL of a 0.1 M iodine solution in methanol (MeOH) (15.7 µmol; 3 eq.) to the solution. Stir the reaction at room temperature for 3-52 h, i.e., until the oxidation is completed.
- Monitor the oxidation reaction via HPLC and MS. Confirm the formation of the second disulfide bond by iodoacetamide (IAA) derivatization (Step 6).
- Stop the reaction by adding 79 µL of a 1 M ascorbic acid solution in H2O (78.8 µmol; 15 eq.). Freeze-dry the reaction mixture and use the powder for the 3rd oxidation.
- 3rd Oxidation (Figure 1D)
NOTE: The last oxidation leads to the deprotection of the tBu-protected cysteines and to the formation of the third disulfide bridge. The protocol applies to 15 mg of peptide after the 2nd oxidation (2718.3 g/mol; 5.5 µmol; 1 eq.).- Dissolve the peptide (15 mg, final concentration of 1 mM) in 5.5 mL of TFA. It contains a scavenger mixture consisting of 11.2 mg of diphenylsulfoxide (55 µmol; 10 eq.), 60.2 µL of anisole (0.6 mmol; 100 eq.) and 97.2 µL of trichloromethylsilane (0.8 mmol; 150 eq.). Stir the mixture for 3-5 h at room temperature.
- Monitor the oxidation reaction via HPLC and MS. Confirm the formation of the third disulfide bond by iodoacetamide (IAA) derivatization (Step 6).
- Precipitate the peptide in the tubes containing cold diethyl ether (0 °C, 1 mL of reaction solution per 8-10 mL of diethyl ether).
- Centrifuge the suspensions (3400 x g, 1 min), decant the supernatant, and wash the pellets repeatedly (4 times) with 8-10 mL of cold diethyl ether (0 °C). Let the pellets dry on air (3 min).
- Dissolve the pellets in 1 mL of 80% tert-butanol (in H2O), freeze-dry the peptide and store it at -20 °C.
5. Peptide Purification
NOTE: Purify the oxidized peptides by semi-preparative RP HPLC equipped with a C18 column (10 µm particle size, 300 Å pore size, 250 x 22 mm) and a 3.6 mL injection loop. Use a gradient elution system of 0.1% TFA in H2O (eluent A) and 0.1% TFA in MeCN/H2O (9:1, eluent B). Detect the peaks at 220 nm.
- Add 15 mg of the freeze-dried crude product from Step 4.3.5 to a 15 mL tube. Dissolve the crude product in the volume of the HPLC sample loop (e.g., 3.6 mL). Use a mixture of 0.1% TFA in MeCN/H2O (1:1). Vortex until complete dissolution and centrifuge (3400 x g) the solution for 1 min.
- Draw up the 3.6 mL mixture in a 5 mL syringe and inject the sample without any air bubbles into the injection loop. Start the injection into HPLC system. Purify the peptide mixture using a gradient of 0-50% eluent B over 120 min at a flow rate of 10 mL/min.
- Collect the fractions in individual tubes as they appear. After the run is complete, prepare selected fractions for MS and HPLC analysis (Step 6.1-6.2). Freeze-dry all fractions and store them at -20 °C.
- After the run is complete, prepare selected fractions for MS and HPLC analysis (Step 6.1-6.2). Freeze-dry all fractions and store them at -20 °C.
6. Peptide Analytics
- Analytical HPLC
NOTE: Confirm the purity of a peptide by analytical RP HPLC equipped with a C18 column (5 µm particle size, 300 Å pore size, 250 x 4.6 mm) and a 500 µL injection loop. Use a gradient elution system of 0.1% TFA in H2O (eluent A) and 0.1% TFA in MeCN (eluent B). Detect the peaks at 220 nm.- Transfer a sample of the peptide fractions or reaction controls into an HPLC vial and dissolve it in a mixture of 0.1% TFA in MeCN/H2O (1:1, 300-500 µL). Place the HPLC vial into the autosampler of the analytical RP HPLC.
- Inject 250 µL of each sample. Use a gradient elution system of 0.1% TFA in H2O (eluent A) and 0.1% TFA in MeCN (eluent B). Purify the peptide using a gradient of 10-40% eluent B over 30 min at a flow rate of 1.0 mL/min.
- MALDI TOF mass spectrometry
NOTE: Confirm the identity of a peptide by MALDI TOF (time of flight) mass spectrometry using α-cyano-4-hydroxycinnamic acid as matrix.- Transfer a visible amount of the peptide into a 1.5 mL microcentrifuge tube and dissolve it in 10 µL of a 37 mM α-cyano-4-hydroxycinnamic acid solution in a mixture of 0.1% TFA in H2O/MeCN (1:1).
- Vortex the solution for 10 s, apply 2 µL of the sample to a ground steel target, and air-dry the sample.
- Use the reflector mode for the measurements and a peptide calibration standard for molar masses below 6000 g/mol.
- Iodoacetamide derivatization
NOTE: As iodoacetamide reacts with thiol groups, iodoacetamide derivatization of the peptides indicates free thiol groups. Hence, the absence of free thiol groups serves as a reaction control via MS during the 1st oxidation.- Dissolve the peptide in 10 mM phosphate buffer (100 µL; 0.05 mM; pH 7.8) in a 1.5 mL microcentrifuge tube. Add 100 µL of iodoacetamide in 10 mM phosphate buffer (4 mM) to the peptide solution and gently shake the reaction for 2 h at room temperature in the dark. Freeze-dry the reaction mixture and store it at -20 °C.
- Use a C18-concentration filter pipette tip and condition the tip with 10 µL of 80% (3 times), 50% (3 times), 30% (3 times) and 0% (3 times) MeCN in H2O (+ 0.1% TFA).
- Dissolve the sample from Step 6.3.1 in 1 µL of 0.1% TFA in H2O and add the solution to the filter pipette tip. Pipette carefully up and down so that the peptide binds to the bead. Remove the H2O out of the pipette tip and rinse the filter pipette tip with 10 µL of 0.1% TFA in H2O.
- Add 2 µl of 0.1% TFA in H2O/MeCN (1:1) with another pipette tip (without filter) on top of the bead which contains the peptide. Apply the filtrate to the ground steel target and air-dry the sample.
- Apply 1 µL of a 37 mM α-cyano-4-hydroxycinnamic acid solution in a mixture of 0.1% TFA in H2O/MeCN (1:1) to the ground steel target with the sample and air-dry the sample.
- Use the reflector mode for the measurements and a peptide calibration standard for molar masses below 6000 g/mol.
- Amino acid analysis
NOTE: Analyze the exact peptide concentration as well as the amino acid composition of the peptide using an amino acid analyzer.- Transfer 100 µg of the pure peptide (2604 g/mol; 0.04 µmol) to a 1.5 mL microcentrifuge tube and dissolve the powder in 200 µL of 6 M HCl.
- Transfer 200 µL of the solution into a glass ampoule and rinse the 1.5 mL microcentrifuge tube twice with 200 µL of 6 M HCl. Transfer the rinsing solution into the glass ampoule as well.
- Close the ampoule by heating the neck of the ampoule with a Bunsen burner flame. Put the ampoule into a glass tube. Place it in a heating block for 24 h at 110 °C for hydrolysis.
- Open the ampoule and transfer the solution into a 2 mL microcentrifuge tube. Wash the ampoule (3 x 200 µL) and the cap (3 x 100 µL) with double-distilled H2O and transfer it into the microcentrifuge tube.
- Centrifuge the solution for 6 h at 60 °C and 210 x g in a rotational vacuum concentrator. Dissolve the hydrolyzed product in 192 µL of the sample dilution buffer (200 µM) and transfer the solution into a micro-centrifugal filter.
- Centrifuge the sample for 1 min at 2300 x g and transfer 100 µL of the filtrate into an amino-acid analysis sample tube. Place the tube into the amino acid analyzer and start the analysis. An amino acid standard is used for calibration.
7. MS/MS Analysis of Disulfide Connectivity
- Partial reduction and alkylation17
- Dissolve 600 µg of the pure peptide (2604 g/mol; 0.23 µmol) in 1.2 mL of 0.05 M citrate buffer (pH 3.0; 0.14 mM peptide concentration) containing 7.5 mg of tris(2-carboxyethyl)phosphine (TCEP; 0.02 M; 0.03 mmol).
- Incubate the mixture at room temperature and take several reaction control samples (100 µL) ranging from 0 min to 30 min.
- Mix the samples in a 1.5 mL microcentrifuge tube with 300 µL of alkylation buffer (0.5 M tris-acetate; pH 8.0; 2 mM ethylenediaminetetraacetic acid (EDTA); 1.1 M iodoacetamide) to stop the reaction and perform carbamidomethylation of the free thiol groups.
- Stop the reaction after 5 min by adding 100 µL of 10% TFA (in H2O) and store the samples on dry ice. Prepare HPLC samples as described in Step 6.1.2 and inject 400 µL. (Figure 2A)
- Use a gradient elution system of 0.1% TFA in H2O (eluent A) and 0.1% TFA in MeCN (eluent B). Analyze the peptides using the combination of an isocratic separation (10 % eluent B for 15 min) and then a subsequent gradient of 10-35 % eluent B over 25 min at a flow rate of 10 mL/min. Detect the peaks at 220 nm.
- Collect the fractions in 1.5 mL microcentrifuge tubes and freeze-dry the peptides. (Figure 2B)
- Transfer a small amount of each fraction to a 1.5 mL microcentrifuge tube for MS/MS analysis of the oxidized forms (continue at Step 7.1.12).
- Dissolve the remaining peptide (10-100 µg) in 0.1% TFA in H2O (10-50 µL) and add an appropriate volume of a 100 mM TCEP solution (in H2O) to get a final TCEP concentration of 10 mM.
- Incubate the reaction for 1 h at 37 °C (Figure 2C). Freeze-dry the reaction mixture and store it at -20 °C.
- Prepare MS/MS samples as described in Step 6.3.2-6.3.5.
- Perform the MS/MS measurements on a MALDI TOF/TOF mass spectrometer. Use MS/MS LID (laser-induced decay) for fragmentation of the peptide and select the precursor masses of 2- and 4-times carbamidomethylated species. Process and evaluate the MALDI data to confirm the desired disulfide connectivity.
8. NMR Experiments and Structure Analysis
- Dissolve approx. 0.3-2 mg of the pure peptide product in 500 µL of H2O/D2O (90:10) and transfer the mixture into an NMR microtube.
- Prepare the sample for measurements at an NMR spectrometer
- Record 2-dimensional [1H,1H]-DQF-COSY (double quantum filtered correlation spectroscopy), [1H,1H]-TOCSY (total correlated spectroscopy), [1H,1H]-NOESY (nuclear Overhauser enhancement spectroscopy), and [1H,13C]-HSQC (heteronuclear single quantum coherence) spectra using water suppression.
- Assign the proton resonances of the recorded spectra and create the atom assignment from NOESY spectra. Compare the intensities in the NOESY spectra to set the upper-limit distance constraints between different atoms in the peptide. Use the intensity of the germinal protons for peak-intensity calibration.
- Perform structure calculation and refinement with a computer program for molecular visualizing and use the identified disulfide connectivities of Step 7 as additional restraints (Figure 3).
- Select the structures with the lowest energies and use it for molecular dynamics (MD) simulations.
Representative Results
15 different disulfide-bridged isomers of the µ-conotoxin PIIIA are synthesized and characterized in detail (Figure 1). Disulfide bonds are identified by partial reduction and subsequent MS/MS analysis (Figure 2). NMR analysis of the different isomers is carried out (Figure 3) to reveal the individual peptide structures. Notably, a combination of RP HPLC, MS/MS fragmentation, and NMR analysis is required for unambiguous identification of the disulfide connectivity.
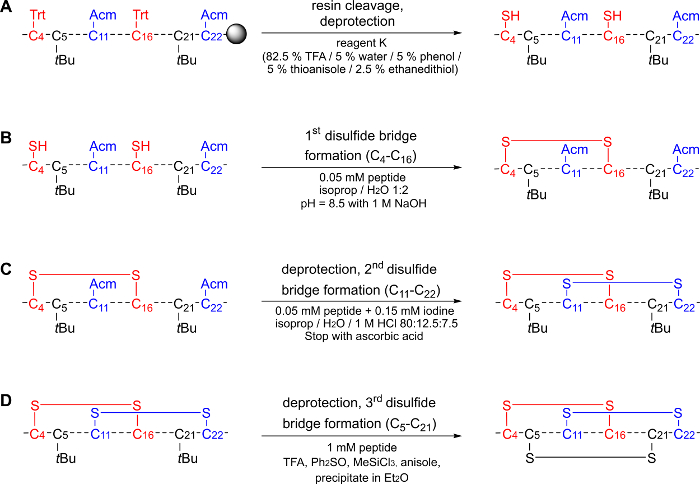
Figure 1: The selective disulfide bond formation of native µ-PIIIA via the protecting group strategy. (A) The deprotection of the Trt-protected cysteines during the peptide cleavage from the resin. (B) The first disulfide bridge formation of the unprotected cysteines. (C) Deprotection and disulfide bridge formation of the Acm protected cysteines. (D) Deprotection and disulfide bridge formation of the tBu protected cysteines. Please click here to view a larger version of this figure.
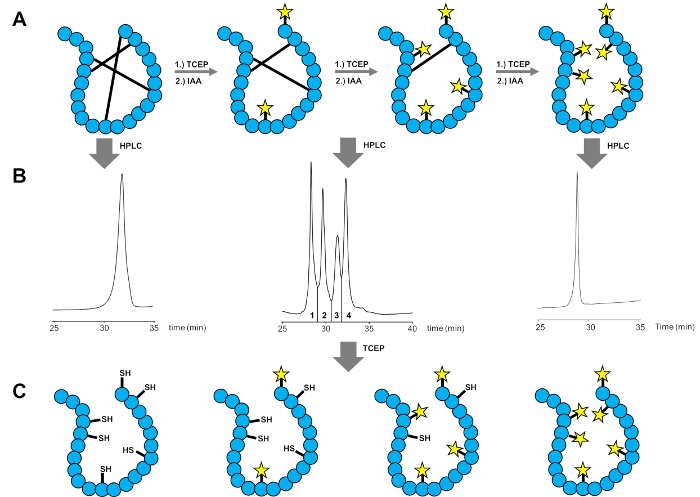
Figure 2: Partial reduction workflow for the disulfide bond assignment by MS/MS Analysis. (A) Partial reduction and alkylation of the peptide. (B) HPLC purification of different reaction control samples. (C) Reduction of the purified samples. Partially carbamidomethylated species (two peptides in the middle) harbor information on the disulfide connectivity which is determined by MS/MS analysis. This figure has been adapted with permission from Heimer, P. et al. Conformational µ-Conotoxin PIIIA Isomers Revisited: Impact of Cysteine Pairing on Disulfide-Bond Assignment and Structure Elucidation. Analytical Chemistry. 90 (5), 3321-3327 (2018). Copyright (2018) American Chemical Society. Please click here to view a larger version of this figure.
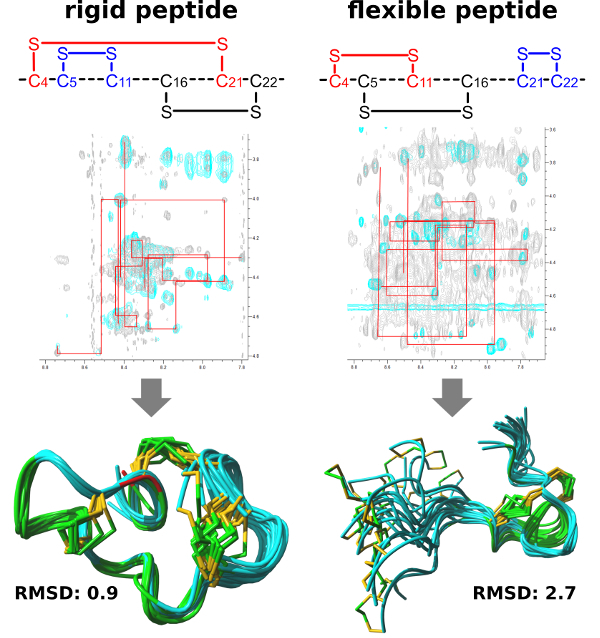
Figure 3: The sequential walk and the resulting NMR solution structure of a rigid (left) and a flexible (right) µ-PIIIa isomer. The 20 structures with the lowest energies as well as the disulfide connectivity of the different isomers are shown. The comparison of the root mean square deviation (RMSD) values clarifies that a rigid peptide mostly leads to a better resolved NMR structure. Please click here to view a larger version of this figure.
Discussion
The method described herein for the synthesis of cysteine-rich peptides such as µ-PIIIA represents a possibility to selectively produce disulfide-bonded isomers from the same amino acid sequence. Therefore, established methods such as Fmoc-based solid phase peptide synthesis18 and a defined protecting group strategy for the regioselective formation of disulfide bonds were used16. The solid-phase peptide synthesis can produce amino acid sequences on a polymer support (resin) via automated synthesis. These amino acids are protected against undesired side reactions during sequence assembly by special protecting groups at their side chains, which are – depending on the protocol used – deprotected upon peptide cleavage from the resin. This protection also applies to the cysteine side chains within the peptide chain, e.g., trityl protection. However, distinct protecting groups for individual pairs of cysteines such as acetamidomethyl and tert-butyl are not concomitantly cleaved through this elimination step. These protecting groups can be cleaved off under conditions which allow subsequent oxidation by forming disulfide bonds from the deprotected thiol groups. In this way, all 15 disulfide isomers of a peptide containing six cysteine residues can be selectively formed7,16. The limiting factor, however, is that with an increasing number of different orthogonal protecting groups the synthetic effort increases, which has a high impact on the yield of the desired disulfide bridged peptide. Thus, in selected cases and in particular for less complex peptides possessing only one or two disulfide bonds the oxidative self-folding approach indeed might be preferred over the protecting group strategy.
Herein, the Trt groups of one cysteine pair are removed by the action of TFA after completion of the peptide assembly and final Fmoc-deprotection of the N-terminal amino acid. The acidic conditions, however, leave the Acm and tBu protecting groups at the other two pairs of cysteine residues intact. Subsequently, purification of the linear peptide containing two unprotected cysteines is performed using preparative HPLC under slightly acidic conditions to maintain the thiol groups in the protonated form. The protocol continues with the first oxidation step after analytical HPLC and MS analysis verified successful synthesis and deprotection of Trt at the cysteine pair of interest. The further steps of deprotection and oxidation of the second and third disulfide bond are carried out and confirmed in the same way using the appropriate protocol for Acm and tBu, respectively. These oxidation reactions are thus simple wet-chemical reactions performed in solution which do not require expensive reagents. The disadvantage, however, is that certain reactions, i.e., connections of distinct cysteine residues, do not proceed smoothly and completely leading to by-products of scrambled disulfide connectivity. Since these are not removed after completion of the individual oxidation reactions, such by-products can accumulate in the crude product mixture. Some of these may possess similar physicochemical properties as the actual peptide, e.g., HPLC elution, which increases the effort for purification of the correctly folded peptide. Although the synthesis and purification could be difficult, as is the case for µ-PIIIA, this method can successfully be used, yet requires good manual and observation skills. Finally, it needs to be considered that every peptide sequence is different and therefore might be treated differently in order to be successful in generating the correct disulfide connectivity.
In addition to the challenging synthesis, it is essential to verify whether the disulfide bonds produced are correct, i.e., represent the intended version of the respective disulfide isomer. This is performed herein using a combination of MALDI MS/MS analysis and NMR structure elucidation. MS/MS analysis is carried out using different partially reduced and alkylated derivatives (carbamidomethylation) of the fully oxidized peptide17. In the MALDI MS/MS LID TOF/TOF spectra an even number of carbamidomethylated cysteines is always found, i.e., 2-, 4- or 6-times carbamidomethylated. This can be explained by the stepwise reduction of the completely oxidized peptides, since in each occasion two thiol functions per disulfide bond are always reduced (opened) and in situ alkylated using iodoacetamide. This carbamidomethylation of two cysteines each can be detected in the MALDI MS/MS spectra and, in turn, refers to the respective disulfide bond these cysteines were engaged in in the intact peptide. For example, the occurrence of four carbamidomethylated cysteines in a sequence with three disulfide bonds helps to identify one specific bond, namely the one formed between the two non-alkylated cysteines, which were not opened during the reaction time of the partial reduction sufficient to open the other two bonds. Thus, by MALDI MS/MS analysis of the various 2- and 4-times carbamidomethylated derivatives of the peptide disulfide isomer, the disulfide connectivities can be completely elucidated and confirmed.
Another possibility to elucidate the disulfide bond pattern is the MS/MS analysis of enzymatically digested peptides, where the peptide has to be proteolytically cleaved at distinct amino acids to produce smaller fragments. These short fragments can still be linked via disulfide bonds, which is the reason that the disulfide pattern can be elucidated from MS/MS analysis of these linked fragments. In case of µ-PIIIA, however, this strategy could not be applied because some cysteines of the sequence are directly adjacent to each other and therefore the digestion of the peptides does not separate the cysteines from each other. Identification of the specific disulfide bridge is therefore difficult.
The structure elucidation by 2D NMR analysis is clearly facilitated in case of a known disulfide connectivity, because this knowledge enables the assignment of nuclear Overhauser effect (NOE's) to specific protons, i.e., referring to the spatial distance between two atoms determined from the intensities of the NOESY signals (through-space NMR experiment). The analysis, however, starts with the sequential walk19 applied to the COSY, TOCSY and NOESY spectra, in this way, it is possible to assign a specific signal to the corresponding H-atom of the amino acids (spin system) in the sequence. The aforementioned distance restraints from the NOESY experiment are used in the structure calculation of the peptide7. The more signals identified, the more accurate the structure will be. However, the difficulty of this assignment increases with the number of amino acids in the peptide and with the occurrence of adjacent cysteines, as the probability increases that signals of several atoms overlap and can no longer be precisely assigned due to close proximity. Furthermore, the flexibility of the peptide chain is decisive for whether signals in the NMR are easily or hardly identifiable. The more flexible a peptide region is, the more conformational changes are occurring, allowing multiple signals to be obtained for one and the same atom. Thus, the intensity decreases with the number of conformations, which causes signals to disappear into background noise. This means that three-dimensional structure elucidation via NMR becomes much easier if the sequence is conformationally constrained.
Finally, this protocol makes it possible to generate multiple disulfide-bridged peptides by monitoring the disulfide bond formation by MALDI MS/MS analysis and concomitant 2D NMR structure elucidation7.
Divulgations
The authors have nothing to disclose.
Acknowledgements
We would like to thank A. Resemann, F. J. Mayer, and D. Suckau from Bruker Daltonics GmbH Bremen; D. Tietze, A. A. Tietze, V. Schmidts and C. Thiele from the Darmstadt University of Technology; O. Ohlenschläger from the FLI Jena, M. Engeser from the University of Bonn; K. Kramer, A. Harzen, and H. Nakagami from the Max Planck Institute for Plant Breeding Research, Cologne; Susanne Neupert from the Institute for Zoology, Cologne; and the Biomolecular Magnetic Resonance Spectroscopy Facilities of the University of Frankfurt for technical support, training modules, and access to instruments. Financial support by the University of Bonn to D.I. is gratefully acknowledged.
Materials
| Fmoc Rink amide resin | Novabiochem | 855001 | |
| Pyr(Boc) | Bachem | A-3850 | |
| Arg(Pbf) | Iris Biotech | FSC1010 | |
| Asn(Trt) | Bachem | B-1785 | |
| Asp(tBu) | Iris Biotech | FSP1020 | |
| Hyp(tBu) | Iris Biotech | FAA1627 | |
| Lys(Boc) | Bachem | B-1080 | |
| Ser(tBu) | Iris Biotech | FSC1190 | |
| Gln(Trt) | Iris Biotech | FSC1043 | |
| Glu(tBu) | Iris Biotech | FSP1045 | |
| Trp(Boc) | Iris Biotech | FSC1225 | |
| Tyr(tBu) | Sigma Aldrich | 47623 | |
| Thr(tBu) | Iris Biotech | FSP1210 | |
| His(Trt) | Iris Biotech | FDP1200 | |
| 2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluorphosphat | Sigma Aldrich | 8510060 | Flammable |
| DMF | Fisher Scientific | D119 | Flammable, Toxic |
| DCM | Fisher Scientific | D37 | Carcinogenic |
| Piperidine | Alfa Aesar | A12442 | Flammable, Toxic, Corrosive |
| N-Methyl-Morpholin | Sigma Aldrich | 224286 | |
| Cys(Acm) | Iris Biotech | FAA1506 | |
| Cys(Trt) | Bachem | E-2495 | |
| Cys(tBu) | Bachem | B-1220 | |
| trifluoruacetic acid | Sigma Aldrich | 74564 | Toxic, Corrosive |
| phenol | Merck | 1002060 | Toxic |
| thioanisol | Alfa Aesar | A14846 | |
| ethanedithiol | Fluka Analytical | 2390 | |
| diethyl ether | VWR | 100,921 | Flammable |
| tert-butanol | Alfa Aesar | L12338 | Flammable |
| acetonitrile | Fisher Scientific | A998 | Flammable |
| water | Fisher Scientific | W5 | |
| isopropanol | VWR | ACRO42383 | Flammable |
| sodium hydroxide | AppliChem | A6579,1000 | Corrosive |
| iodoacetamide | Sigma Aldrich | I6125 | |
| iodine | Sigma Aldrich | I0385 | |
| Hydrochloric acid | Merck | 110165 | Corrosive |
| ascorbic acid | Sigma Aldrich | A4403 | |
| diphenylsulfoxide | Sigma Aldrich | P35405 | |
| anisol | Sigma Aldrich | 96109 | Flammable |
| trichloromethylsilane | Sigma Aldrich | M85301 | Flammable |
| sample dilution buffer | Laborservice Onken | ||
| sodium dihydrogen phosphate | Sigma Aldrich | 106370 | |
| disodium hydrogen phosphate | Sigma Aldrich | 795410 | |
| (2-carboxyethyl)phosphine hydrochloride | Sigma Aldrich | C4706 | |
| citric acid | Sigma Aldrich | 251275 | |
| sodium citrate dihydrate | Sigma Aldrich | W302600 | |
| tris-acetate | Carl Roth, | 7125 | |
| Ethylenediaminetetraacetic acid | Sigma Aldrich | E26282 | |
| peptide calibration standard II | Bruker Daltonics GmbH | 8222570 | |
| Name of Equipment | Company | ||
| solid-phase peptide synthesizer | Intavis Bioanalytical Instruments AG | EPS 221 | |
| lyophilizer | Martin Christ GmbH | Alpha 1-2 Ldplus | |
| semipreparative HPLC | Jasco | system PV-987 | |
| Eurospher 100 C18 column (RP, 5 µm particle size, 100 Å pore size, 250 x 32 mm) | Knauer | 25QE181E2J | purification of the linear peptide |
| Vydac 218TP1022 column (RP C18, 10 µm particle size, 300 Å pore size, 250 x 22 mm) | Hichrom-VWR | HICH218TP1022 | purification of the oxidized peptide |
| analytical HPLC | Shimadzu | system LC-20AD | |
| Vydac 218TP54 column (C18 RP, 5 µm particle size, 300 Å pore size, 250 x 4.6 mm) | Hichrom-VWR | HICH218TP54 | analytical column |
| ground steel target (MTP 384) | Bruker Daltonics GmbH | NC0910436 | MALDI preparation |
| C18-concentration filter (ZipTip) | Merck KGaA | ZTC18S096 | MALDI preparation |
| MALDI mass spectrometer | Bruker Daltonics GmbH | ultraflex III TOF/TOF | |
| amino acid analyzer | Eppendorf-Biotronik GmbH | LC 3000 system | |
| NMR spectrometer Bruker Avance III | Bruker Daltonics GmbH | Bruker Avance III 600 MHz | |
| computer program for molecular visualising | YASARA Biosciences GmbH | Yasara structures | NMR structure calculation |
| computer program for MALDI data evaluation | Bruker Daltonics GmbH | flexAnalysis, BioTools | MS/MS fragmentation |
| analog vortex mixer | VWR | VM 3000 | |
| Microcentrifuge | Eppendorf | 5410 | |
| Centrifuge | Hettich | EBA 20 | |
| Rotational vacuum concentrator | Christ | 2-18 Cdplus | |
| Analytical Balance | A&D Instruments | GR-202-EC |
References
- Fosgerau, K., Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discovery Today. 20 (1), 122-128 (2015).
- Gongora-Benítez, M., Tulla-Puche, J., Albericio, F. Multifaceted roles of disulfide bonds. peptides as therapeutics. Chemical Reviews. 114 (2), 901-926 (2014).
- Tietze, A. A., et al. Structurally diverse µ-conotoxin PIIIA isomers block sodium channel NaV1.4. Angewandte Chemie – International Edition. 51 (17), 4058-4061 (2012).
- Carstens, B. B., et al. Structure-Activity Studies of Cysteine-Rich α-Conotoxins that Inhibit High-Voltage-Activated Calcium Channels via GABABReceptor Activation Reveal a Minimal Functional Motif. Angewandte Chemie – International Edition. 55 (15), 4692-4696 (2016).
- Zhang, Y., Schulten, K., Gruebele, M., Bansal, P. S., Wilson, D., Daly, N. L. Disulfide bridges: Bringing together frustrated structure in a bioactive peptide. Biophysical Journal. 110 (8), 1744-1752 (2016).
- Nielsen, K. J., et al. Solution structure of µ-conotoxin PIIIA, a preferential inhibitor of persistent tetrodotoxin-sensitive sodium channels. Journal of Biological Chemistry. 277 (30), 27247-27255 (2002).
- Heimer, P., et al. Conformational µ-Conotoxin PIIIA Isomers Revisited: Impact of Cysteine Pairing on Disulfide-Bond Assignment and Structure Elucidation. Analytical Chemistry. 90 (5), 3321-3327 (2018).
- Chang, J. Y. Diverse pathways of oxidative folding of disulfide proteins: Underlying causes and folding models. Biochimie. 50 (17), 3414-3431 (2011).
- Böhm, M., et al. Novel insights into structure and function of factor XIIIa-inhibitor tridegin. Journal of Medicinal Chemistry. 57 (24), 10355-10365 (2014).
- Postma, T. M., Albericio, F. Disulfide Formation Strategies in Peptide Synthesis. European Journal of Organic Chemistry. 2014 (17), 3519-3530 (2014).
- Albericio, F., Isidro-llobet, A., Mercedes, A. Amino Acid-Protecting Groups. Chemical Reviews. 109 (6), 2455-2504 (2009).
- Annis, I., Hargittai, B., Barany, G. Disulfide bond formation in peptides. Methods in Enzymology. 289 (1988), 198-221 (1997).
- Kamber, B., et al. The Synthesis of Cystine Peptides by Iodine Oxidation of S-Trityl-cysteine and S-Acetamidomethyl-cysteine Peptides. Helvetica Chimica Acta. 63 (4), 899-915 (1980).
- Bosch, D. E., Zielinski, T., Lowery, R. G., Siderovski, D. P. Evaluating Modulators of Regulator of G-Protein Signaling (RGS) Proteins. Current Protocols in Pharmacology. 56 (2.8), 1-15 (2012).
- Mochizuki, M., Tsuda, S., Tanimura, K., Nishiuchi, Y. Regioselective formation of multiple disulfide bonds with the aid of postsynthetic S-tritylation. Organic Letters. 17 (9), 2202-2205 (2015).
- Peigneur, S., et al. δ-conotoxins synthesized using an acid-cleavable solubility tag approach reveal key structural determinants for NaV subtype selectivity. Journal of Biological Chemistry. 289 (51), 35341-35350 (2014).
- Heimer, P., et al. Application of Room-Temperature Aprotic and Protic Ionic Liquids for Oxidative Folding of Cysteine-Rich Peptides. ChemBioChem. 15 (18), 2754-2765 (2014).
- Kates, S. A., Albericio, F. . Solid-Phase Synthesis: A Practical Guide. , (2000).
- Wüthrich, K. NMR studies of structure and function of biological macromolecules (Nobel lecture). Angewandte Chemie – International Edition. 42 (29), 3340-3363 (2003).
