1. Pretreatment of the Walnut Feedstock before Lignin Extraction
- Production of cut walnut shells
- Feed the walnut shells to a hammer cutter to fracture the shells. Equip the hammer cutter with a 5 mm sieve at the outlet. Collect the fractured walnut shells in a 1 L glass beaker.
- Feed the fractured shells to a micro hammer cutter to obtain ground shells. Equip the micro hammer cutter with a 2 mm sieve at the outlet. Collect the ground walnut shells in a 1 L glass beaker.
- Extraction of fatty acids from ground walnut shells
- Put 150 g of the cut walnut shells in a 500 mL round-bottom flask. Add 200 mL of toluene and a stirring bar to the round-bottom flask.
- Attach a reflux condenser to the round-bottom flask. Heat the mixture at reflux temperature (111 ˚C) with an oil bath for 2 h with vigorous stirring.
- Stop the heating after 2 h and let the mixture cool down to room temperature by removing it from the oil bath.
- Remove the toluene by filtration (185 mm diameter, 10 µm pore size). Discard the toluene filtrate.
- Remove the toluene residues by heating the walnut shells overnight in a vacuum oven at 80 ˚C and 50 mbar.
- Milling of pre-extracted walnut shells
- Place 7 ZrO2 grinding balls with a 20 mm diameter into a 250 mL grinding bowl made of ZrO2.
- Fill the bowl with 40 g of walnut particles. Add 60 mL of isopropanol to the grinding bowl.
- Perform the grinding of the walnut shells with a rotary ball mill. Grind in 4 cycles of 2 min grinding at 27 x g followed by a 4 min pause. Keep the temperature of the bowl below 80 ˚C at any time. Perform no more than 3 batches and let the bowl cool down afterwards.
- Collect the finely ground walnut shells into a 500 mL round-bottom flask. Remove the isopropanol by rotary evaporation at 40 ˚C and 125 mbar.
- Dry the walnut shells overnight in a vacuum oven at 50 ˚C and 50 mbar.
- Sieve the finely ground walnut shells through a 1 mm sieve. Ground the particles that are too large again with the rotary ball mill.
2. Preparation of the Wood Feedstocks
- Cutting of the wooden planks
- Place the wooden planks under a drill, which is equipped with a flat wood speed drill bit. Collect the wood shavings in a glass beaker.
- Place the wooden shavings in a coffee grinder to cut them into smaller pieces.
- Extraction of fatty acids from wood
- Perform the extraction of the fatty acids from wood in precisely the same manner as described for walnut shells in Step 1.2.
NOTE: No milling of the wood is performed in the ball mill, as the conditions described in Step 1.3 did not result in a reduction of the particle size.
- Perform the extraction of the fatty acids from wood in precisely the same manner as described for walnut shells in Step 1.2.
3. Extraction of High β-O-4 Ethanosolv Lignin
- Mild ethanol extraction (Method A)
- Put 25 g of the feedstock in a 500 mL round-bottom flask. Add an 80:20 ethanol/water mixture (200 mL), 4 mL of 37% HCl solution (0.24 M) and a magnetic stirring bar to the round-bottom flask.
- Attach a reflux condenser to the round-bottom flask. Heat the mixture atreflux temperature with an oil bath for 5 h with vigorous stirring.
- Allow the mixture to cool to room temperature by removing it from the oil bath. Filter the mixture (185 mm diameter, 10 µm pore size) and wash the residue with 4 times 25 mL of ethanol.
- Work-up and isolation of lignin
- Collect the liquor in a 500 mL round-bottom flask. Concentrate the liquor by rotary evaporation at 40 ˚C and 150 mbar.
- Dissolve the obtained solid in 30 mL of acetone. Use an ultrasonic bath if the solid does not dissolve completely.
- Precipitate the lignin by adding the mixture to 600 mL of water. If no precipitation occurs, add a small amount of saturated aqueous Na2SO4 solution to flocculate the lignin.
- Collect the lignin by filtration (185 mm diameter, 10 µm pore size). Wash the lignin with 25 mL of water 4 times. Discard the filtrate if no analysis of the hemicellulose fraction is required. If the filtrate is very turbid, add it to a centrifugation tube and collect the (solid) bottom fraction by centrifugation.
- Allow the lignin to air dry overnight. Dry the lignin further in a vacuum oven (overnight at 50 ˚C and 50 mbar).
- Determine the yield after the lignin is dried overnight in a vacuum oven.
- Determine the lignin extraction efficiency by dividing it with the total lignin content as determined by the Klason method24.
- Higher temperature ethanol extraction (Method B)
- Put 15 g of the feedstock in a 250 mL autoclave. Add an 80:20 ethanol/water mixture (120 mL), 2.4 mL of HCl (0.24 M) and a magnetic stirring bar.
- Heat the mixture at 120 ˚C for 5 h with a stirring speed of 5.2 x g. Cool the mixture afterwards in an ice bath.
- Filter the mixture (185 mm diameter, 10 µm pore size) and wash the residue 4 times with 15 mL of ethanol.
- Perform further work-up and isolation precisely as described in Step 3.2.
- Larger scale higher temperature ethanol extraction of walnut shell (Method C*)
- Put 90 g of finely ground walnut shell in a 1 L high pressure autoclave. Add an 80:20 ethanol/water mixture (750 mL) and 6.25 mL of H2SO4 (0.12 M).
- Heat the mixture at 120 ˚C for 5 h with a stirring speed of 35.8 x g. Cool the mixture back to room temperature by turning on the reactors cooling system after the 5 h reaction time.
- Filter the mixture (185 mm diameter, 10 µm pore size) and wash the residue 4 times with 75 mL of ethanol.
NOTE: Using multiple filters saves a lot of time. - Collect the liquors in 2 equal batches. Perform the work-up and isolation as described as in Step 3.2 with double amount of the solvents for both batches.
- Control experiments of Step 3.1. (Method A*) and Step 3.3. (Method B*) (optional)
- Put the exact some materials as described in Step 3.1.1 but replace the HCl solution with 1.67 mL of H2SO4 (0.12M). The rest of Step 3.1-3.2 is identical to Method A.
- Put the exact some materials as described in Step 3.3.1 but replace the HCl solution with 1.0 mL of H2SO4 (0.12M). The rest of Step 3.3 is identical to Method B.
4. De-etherification of the Lignin (optional)
- Dissolve 1000 mg of lignin in a 24 mL of 1:1 1,4-dioxane/water mixture in a 100 mL round-bottom flask. Add 1 mL of a 37% HCl solution to the mixture.
- Add a stirring bar and attach a reflux condenser to the round-bottom flask. Heat the mixture to 100 ˚C with an oil bath for 5 h with vigorous stirring.
- Allow the mixture to cool down to room temperature by removing it from the oil bath. Add the mixture to 160 mL of water to precipitate the lignin.
- Collect the lignin by filtration (185 mm diameter, 10 µm pore size) and wash the lignin with 25 mL of water 2 times. Allow the lignin to air dry overnight. Dry the lignin further in a vacuum oven (overnight at 50 ˚C and 50 mbar).
5. Analysis of Lignin
- Two-dimensional nuclear magnetic resonance (2D-NMR) analysis
- Dissolve 60 mg of dried lignin in 0.7 mL of d6-acetone. Add a few drops of D2O if the lignin does not fully dissolve. Put the mixture in an NMR-tube and take a 2D proton heteronuclear single quantum coherence spectra (HSQC) with an NMR spectrometer with the following parameters: (11, -1), (160, -10), nt = 4, ni = 51220.
- Analyze the obtained HSQC spectra. Adjust the spectra by manual phase corrections on both axis until all signals are positive, as this is especially important along the horizontal (f2) axis. Perform no baseline corrections. The positions of all the linkages are given in Step 5.1.3 and 5.1.6.
- Integrate the signals in the aromatic region that correspond to the three different aromatic units (proton numbering as per Figure 4). These signals are in the region [(Proton range)(Carbon range)]:
S2/6: [(6.48-6.90)(104-109)]
S'2/6: [(7.17-7.50)(105-109)]
Scondensed: [(6.35-6.65)(106-109)]
G2: [(6.78-7.14)(111.5-116)]
G5: [(6.48-7.06)(115-120.5)]
G6: [(6.65-6.96)(120.5-124.5)]
H2/6: [(7.05-7.29)(128.5-133)]
Note: H3/5 overlaps with the G5 signal, and it is assumed that H2/6 has the same intensity as H3/5. The signal for condensed G overlaps with G5. If no (or hardly any) G2 and G6 signals are present, this indicates that full condensation of G has occurred. - Calculate the amount of aromatic units with the formula:
Total aromatic = (((S2/6 + S'2/6)/ 2) + Scondensed) + ((G2 + G5 + G6– H2/6) / 3) + (H2/6 / 2) - Calculate the percentage of G, H and S units with the following formulas:
Ratio S = (((S2/6 + S'2/6)/ 2) + Scondensed): total aromatic x 100%
Ratio G = ((G2 + G5 + G6– H2/6) / 3) : total aromatic x 100%
Ratio H = (H2/6 / 2) : total aromatic x 100% - Integrate the signals in the aliphatic region signals that correspond to the β-O-4, β-β and β-5 linkages and Hibbert Ketones. These are in the region [(proton range)(carbon range)]:
β-O-4α [(4.76-5.10)(73-77.5)]
β'-O-4α [(4.44-4.84)(81.5-86)]
β-O-4β and β'-O-4β [(4.03-4.48)(85-90.5)]
β-O-4γ and β'-O-4γ [(3.10-4.00)(58.5-62)]
β-5α [(5.42-5.63)(88-92)]
β-5β [(3.36-3.56)(53-54.5)]
β-5γ [(3.50-4.00)(62-64.5)]
β-βα [(4.59-4.77)(86.5-89.5)]
β-ββ [(2.98-3.20)(55.5-59)]
β-βγ [(3.75-3.96)(72.5-76)] and [(4.10-4.31)(72.5-76)]
HKγ [(4.20-4.30)(66-68)]
Note: The β-protons of the β-O-4 and β'-O-4 linkages overlap. The structural motifs of these linkages are given in Figure 1. - The total number of linkages per 100 C9 units are all based on the signal of the α proton of the linkages. Calculate the total number of linkages with the following formulas:
β-O-4 linkages = (β-O-4α + β'-O-4α) / total aromatic x 100
# β-5 linkages = β-5α / total aromatic x 100
β-βlinkages = β-βα / total aromatic x 100
- Gel permeation chromatography (GPC) analysis
- Dissolve 10 mg of dried lignin in 1 mL of tetrahydrofuran (THF) (with a drop of toluene as the internal standard). Filter this mixture through a 0.45 µm syringe filter into an autosampler vial with a reduced volume inlet of 0.3 mL. Close the autosampler vial with a cap.
- Inject 20 µL of the sample into a THF GPC. Process the obtained data.
- Correct the obtained signal for the reference signal (toluene). Select the elution volume for the correct range (~200-10000 Da). Calculate mass distribution by the software.
6. Depolymerization of Lignins to Phenolic 2-Arylmethyl-1,3-Dioxolanes (Acetals)
- Place 50 mg of dried lignin in a 20 mL microwave vial as the reaction vessel equipped with a magnetic stirrer. Add 0.85 mL of 1,4-dioxane, 50 µL of ethylene glycol in 1,4-dioxane (0.54 mL/mL) and 50 µL of octadecane (internal standard) in 1,4-dioxane (26 mg/mL).
- Close the reaction vessel and heat the solution to 140 °C while stirring at 3.8 x g.
- When the reaction vessel reaches 140 °C, add 50 µL of Fe(III)OTf3 in 1,4-dioxane (0.1 g/mL).
- Stir the reactor for 15 min.
- Cool the reactor to room temperature and remove the depolymerization liquid as described in Step 7.1.
7. Work up and Analysis of Depolymerization Mixtures
- Filter the liquid over Celite (permeability: 2.60-6-50 darcy; particle size: 150 mesh Tyler; sieve retain (140 M US): 2.0-25.0%) and collect in a 2 mL centrifuge tube.
- Concentrate the liquid overnight at 35 °C in a rotational vacuum concentrator.
- Extract the final oil/solid with the following procedure:
- Suspended and swell the residue in 0.15 mL of dichloromethane (DCM) by extensive mixing (by vortex), 15 min of sonication and 30 min in automatic wheel.
- Centrifuge the samples for up to 10 s using a minispin tabletop centrifuge to ensure that the liquid is at the bottom of the tube (centrifugation speed: 671 x g).
- Add 0.75 mL of toluene and mix extensively (by vortex and 10 min sonication).
- Centrifuge the samples for up to 10 s using a minispin tabletop centrifuge (centrifugation speed: 671 x g) to ensure that the liquid is at the bottom of the tube.
- Separate the light organic liquid from the solid or thick oily residue and filter this liquid over a plug of Celite and collect in a glass vial.
NOTE: This procedure for suspension/washing is repeated three times, and in the last extraction, 0.5 mL of toluene is used.
- Concentrate the combined organic phases by rotary evaporation (40 ˚C, 20 mbar).
- Dissolve the oily residue in 1 mL of DCM for gas chromatography flame ionization detector (GC-FID) analysis.
- Perform GC-FID using a GC equipped with an FID detector and use helium as the carrier gas. Standard settings: 1 µL injection, a split ration of 50:1, a helium flow of 0.95 mL/min. Equip the GC apparatus with a HP5 column (30 m x 0.25 mm x 0.25 µm) and run with a temperature profile which starts with a 5 min 60 °C isotherm. Follow-up by a 10 °C/min ramp for 20 min to 260 °C. Hold this temperature for 20 min.
- Integrate the peaks in the spectra manually. The retention times of the peaks are as following: octadecane (21.4 min), H-acetal (19.5 min), G-acetal (20.8 min), S-acetal (23.4 min). Use the obtained values in Step 7.8 to perform the quantification.
- Perform the quantification of G acetal, which is based on a calibration curve with a standard compound isolated using an internal standard (octadecane).
NOTE: Calibration curve: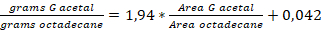 (R2=0,9991)
(R2=0,9991)
Yield G-acetal: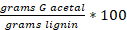
Based on previous results9,21,23 a response factor was estimated for H and S acetal as 2.19 and 1.82, respectively.
In Figure 2, the obtained feedstock after pretreatment are shown (left column). All feedstock was obtained as small chips present apart from beech wood, which was acquired as shavings of suitable particle size for extraction. The lignins obtained after the extraction show a wide range of colors and particle sizes. The lignins obtained from mild treatments (method A and second column Figure 2) are typically red/pink in color and acquired as small flakes. When harsher conditions are applied (methods B and C*), the obtained lignins have a brown/brownish yellow color (third and fourth column Figure 2). The yield did increase for all the extractions performed under harsher conditions (methods B and C*) compared to milder conditions (Reaction scheme in Figure 1, results in Table 1). This effect was much more profound for walnut (10.2% increase), beech (8.5% increase) and cedar wood (5.1% increase) compared to pine wood (only 0.5% increase). Based on the lignin content of the biomass before extraction (40.3% for walnut, 28.6% for pine25, 18.8% for beech25 and 35.1% for cedar25), the lignin extraction efficiency of beech wood is especially high (73.9%), whereas for the other sources lower extraction efficiency was obtained. Methods A* and B*, control experiments with sulfuric acid for methods A and B, showed some clear differences in extraction yield. Mild extraction of walnut shells with sulfuric acid (method A*) gave only a very low yield of 2.6%, which is markedly lower than the extraction with hydrochloric acid (method A) (2.6% and 5.0%). However, with harsher extraction conditions, the extraction with sulfuric acid (method B*) shows a higher yield compared to hydrochloric acid (method B) (19.3% and 15.2%), but it should be noted that sugar traces are present in the product obtained by extraction with sulfuric acid.
From the NMR analysis of the different lignins (example shown in Figure 4), the H/G/S ratio and amount of linkages were determined (Table 1). Due to the overlap of the β and γ-protons of the β-O-4 and the β'-O-4 linkage, the amount of linkages is quantified using the α-protons. Additionally, the G5/6 and H3/5 signals overlap but these can be corrected by adjusting the ratios accordingly using the H2/6 signals. Also, a signal corresponding to the γ-protons of the Hibbert Ketones and a signal for oxidized S units, which likely are caused by lignin end-groups, are identified.
The ratios obtained from NMR show that in general extractions with method B provides lignin with higher S content compared to those with method A in the case that the native material contains S units. Also, the extractions with method B provide lignin with a lower amount of total β-O-4 linkages compared to method A, indicating increased degradation upon increase in temperature. An exception is the walnut lignin obtained from methods methods A and B for which the amount of total β-O-4 linkages was very similar. The number of β-β and β-5 linkages does decrease when harsher conditions are applied, although to a lesser extent. Additionally, NMR revealed that all the lignins obtained after ethanol extraction showed a degree of structural modification of the β-O-4 linkage. These have at least ~50% substitution at the α-OH group, resulting in the α-ethoxylated β'-O-4 linkage. The lignin extraction shows high reproducibility, which was proven by performing the mild extraction of walnut shells (method A) 4 times. Especially, the deviation in the total number of β-O-4 linkages is remarkably small. When the extraction was performed under harsher conditions (methods B and C*), the percentage of α-ethoxylation increased. In the HSQC spectra of beech lignin extracted at harsher condition (method B), a signal for S condensed is visible, which fits perfectly with the significant decrease in the amount of β-O-4 linkages. The walnut extraction performed at large scale (method C*) shows a significant decrease for all linkages and a signal for S condensed is visible in the HSQC spectra. The relatively high yield for the extraction of cedar at mild conditions (method A) is caused by the presence of a substantial amount of fatty acid. Control experiments with sulfuric acid gave good insight in the effect of the acid on the composition of the obtained lignin. With mild extraction conditions (methodA*), a very pure lignin was obtained which was similar in composition compared to the other mild extractions (methodA). The somewhat lower amount of β-O-4 linkages can be attributed to a less efficient incorporation of ethanol into the lignin framework, resulting in a lower number of β'-O-4 linkages. At harsher extraction conditions (method B*), the differences with the obtained lignin is much more profound compared with the lignin obtained from walnut shells extracted in the presence of hydrochloric acid (method B). The total number of β-O-4 linkages shows a sharp decrease (35 and 74, respectively) and the lignin obtained with sulfuric acid shows a high amount of condensation in the aromatic region (48%), which was determined by the integration of the signals corresponding the Scondensed and Gcondensed (Step 5.1.3). This high amount of condensation can be fully attributed to sulfuric acid, as the product obtained from the same extraction with hydrochloric acid showed no condensation in the aromatic region. The composition of the product obtained at harsh larger scale extraction (method C*) shows no big difference with the product obtained at a smaller scale (method B*). The only big difference is the lower amount of condensation in the aromatic region in the large-scale extraction (9%) and subsequently a higher amount of β-O-4 linkages. This difference could be caused by the difference in the heating profile between the different autoclaves.
The lignins were also analyzed by GPC (Figure 5) to provide insight in the molecular weight (Table 2). These reveal that when harsher extraction conditions (method B) are applied, both the weight average molecular weight (Mw) and the polydispersity are increasing for all sources. The number average molecular weight (Mn) between the extraction conditions are comparable for each source. Overall, these results show that harsher extraction conditions have a two-fold effect, and larger fragments are extracted in addition to additional breakdown of such fragments.
For some applications, the formation of β'-O-4 linkage is undesired, for example, when applying depolymerization methods that rely on the oxidation of the benzylic (α) hydroxyl group26,27,28. The transformation of β'-O-4 linkage of ethanosolv lignin to regular β-O-4 linkages was previously reported20 and was performed with a lignin batch obtained from walnut shells that is comparable to the lignin obtained from walnut shells reported in this paper (Figure 6). This lignin consisted of 30 native β-O-4 linkages and 39 α-ethoxylated β'-O-4 linkages (34 and 38 linkages, respectively for the lignin in this paper). De-etherification converted almost all the α-ethoxylated linkages to the native structure as the obtained lignin consisted of 57 β-O-4 linkages and only 3 α-ethoxylated β'-O-4 linkages, showing a small loss in the total number of β-O-4 units. The mass of the lignin was 72% of the original lignin, which is primarily caused by the loss of the ethyl group.
To demonstrate the potential of the lignin for the production of aromatic monomers through mild depolymerization, acidolysis reations with Fe(OTf)3 in the presence of ethylene glycol were performed (Figure 7). This reaction yields three different phenolic 2-arylmethyl-1,3-dioxolanes (acetals) that relate to the H, G and S units present in the lignin. Table 3 shows the yield of the S, G and H acetals and the total yields are shown in Figure 8. It is visible that the lignin extraction method has a major effect being the yield of acetals. Lower yields are obtained for lignin extracted using harsher conditions (method B). This is likely due to a more modified (higher percentage of α-ethoxylation) condensed structure as described in the previous paragraph.
The importance of the β-O-4 units is reflected by providing correlations to monomer yield in depolymerization such as presented in the protocol (Figure 9). A clear trend is visible considering the total β-O-4 content and the non-etherified β-O-4 linkages, where a higher β-O-4 content generally results in higher yield of phenolic 2-arylmethyl-1,3-dioxolanes (acetals) which is in line with previous results21. When considering the etherified β'-O-4 linkages, the trend is also clear, showing that the depolymerization yield is not related to the number of β'-O-4 linkages. Under reaction conditions, the etherified β-O-4 linkages can be de-etherified but this additional step results in the loss of material, as described earlier.
Overall, correcting the monomer depolymerization yield for the lignin extraction yield, the following results can be obtained (Table 4). These show that comparing methods A & B, generally higher amounts of acetal can be obtained by harsher extraction providing higher overall lignin yields followed by a (less selective) depolymerization. Nevertheless, the results for pinewood also show that this is dependent on the biomass source since the increase in extraction severity does not provide a significant yield increase. Retention of the β-O-4 structure is preferred for this wood type to give higher overall phenolic 2-phenylmethyl-1,3-dioxolane (acetals) yields.
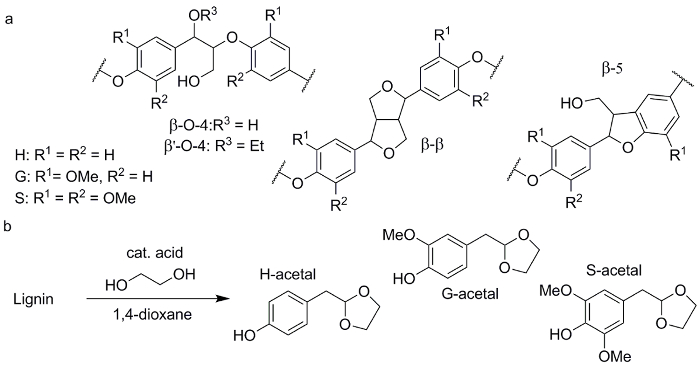
Figure 1. Chemical structures of the obtained products. (a) Common structural motifs as present in the lignin structure. (b) Acid catalyzed lignin depolymerization combined with acetal trapping to obtain phenolic 2-arylmethyl-1,3-dioxolanes (acetals). Please click here to view a larger version of this figure.

Figure 2. Obtained lignin from different feedstocks. Image of the four different lignocellulose feedstocks after pretreatment (Steps 1 and 2) and the obtained lignins after organosolv extraction at different conditions (method A-step 3.1, method B-step 3.3 and method C*-step 3.4). Please click here to view a larger version of this figure.
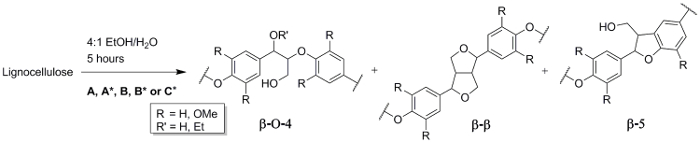
Figure 3. Reaction scheme for ethanosolv extraction. Overview of the obtained linkages: β-O-4 (R' = H), β'-O-4 (R' = Et), β-β and β-5. Conditions: (A) 80 ˚C, 0.24 M HCl (step 3.1), (B) 120 ˚C, 0.24 M HCl (step 3.3), (C*) 120 ˚C, 0.12 M H2SO4 (step 3.4) and the control experiment A* and B* (step 3.5). Please click here to view a larger version of this figure.
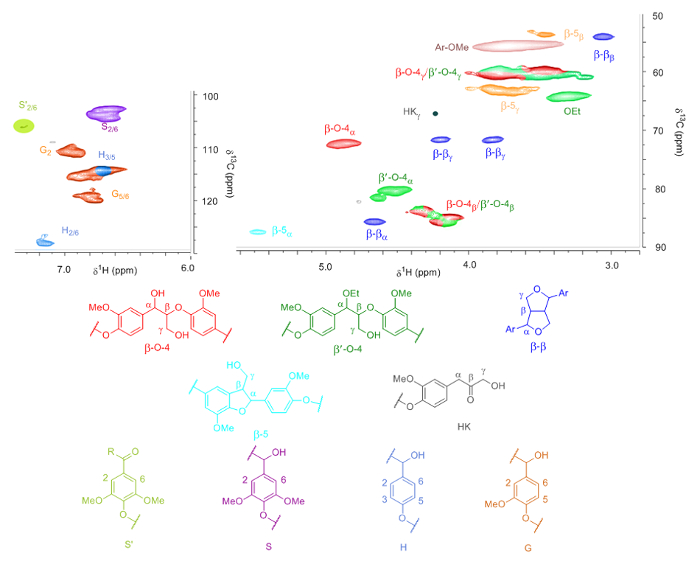
Figure 4. HSQC analysis of lignin. Identification of all lignin linkages measured with 2D-HSQC of lignin obtained from walnut shells using mild treatment (step 3.1). The signals for HKγ and S'2/6 are magnified to make them visible. Please click here to view a larger version of this figure.
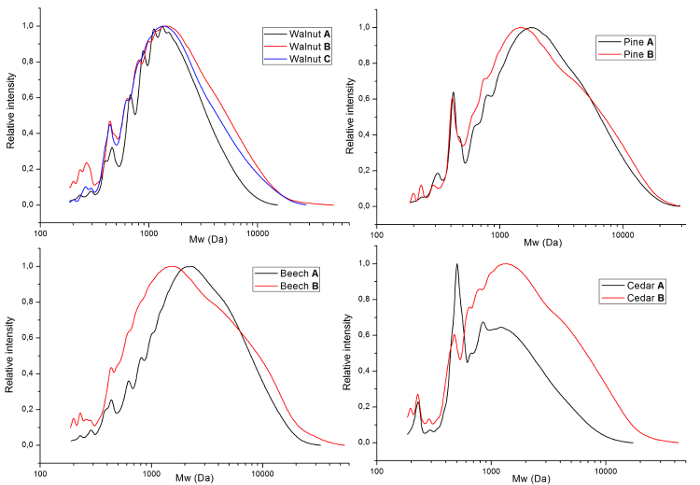
Figure 5. Molecular weight of lignin. GPC graphs of the obtained lignins divided by source (a = walnut, b = pine wood, c = beech wood and d = cedar wood). The lines correspond to different samples as given by Table 2. Please click here to view a larger version of this figure.
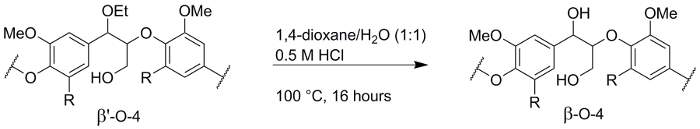
Figure 6. De-etherification of lignin. Reaction scheme of the de-etherification of the obtained ethanosolv lignin from walnut shells (step 4). Please click here to view a larger version of this figure.
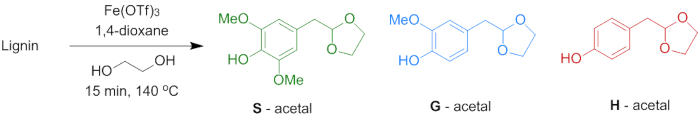
Figure 7. Lignin depolymerization to acetals. Reaction scheme for depolymerization of lignin to phenolic 2-arylmethyl-1,3-dioxolanes (acetals). H unit: R1 = R2 = H; G unit: R1 = OMe, R2 = H; S unit: R1 = R2 = OMe (step 6). Please click here to view a larger version of this figure.
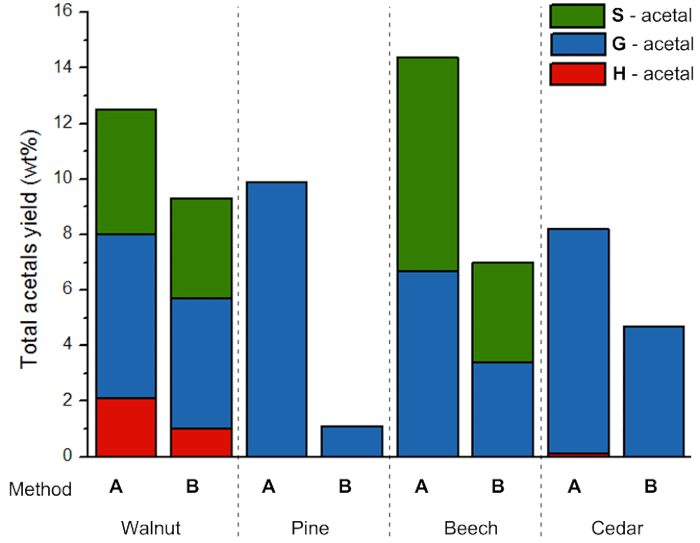
Figure 8. Acetal yield per source. Yields of phenolic 2-arylmethyl-1,3-dioxolanes (acetals) obtained from depolymerization of lignin from different sources. Please click here to view a larger version of this figure.
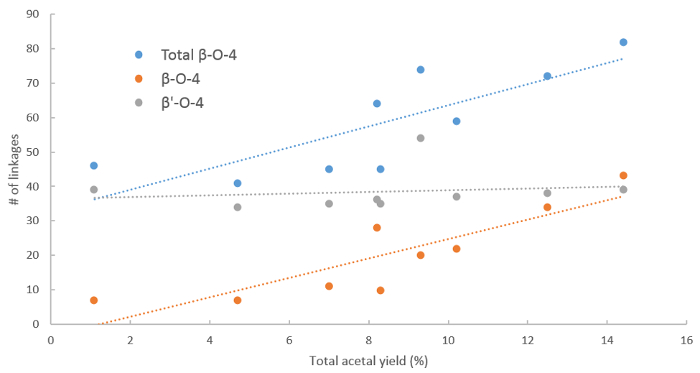
Figure 9. Influence of β-O-4 linkages on the acetal yield. Yields of phenolic 2-arylmethyl-1,3-dioxolanes (acetals) obtained from lignin depolymerization compared to the total β-O-4 (blue), non-etherified β-O-4 (orange) and etherified β-O-4 (gray) content in the lignin feedstock. Please click here to view a larger version of this figure.
| Source | Conditions | Yield (%) | Extraction efficiency (%)1 | S/G/H ratio | Total β-O-4 | β-O-4 | β’-O-4 | β-β | β-5 |
| Walnut | A | 5.0 ± 0.7 | 12.4 | 45/46/9 | 75 ± 2.5 | 36 ± 2.6 | 39 ± 3.1 | 11 ± 0.7 | 5 ± 1.5 |
| Walnut | A* | 2.6 | 6.5 | 47/45/8 | 53 | 32 | 21 | 9 | 4 |
| Walnut | B | 15.2 | 37.7 | 59/37/4 | 74 | 20 | 54 | 9 | 6 |
| Walnut2 | B* | 19.3 | 47.9 | 75/25/0 | 35 | 5 | 30 | 7 | 3 |
| Walnut2 | C* | 16.2 | 40.2 | 65/33/2 | 45 | 10 | 35 | 8 | 3 |
| Pine | A | 3.5 | 12.2 | 0/>99/<1 | 59 | 22 | 37 | 0 | 14 |
| Pine | B | 4.0 | 14.0 | 0/>99/<1 | 46 | 7 | 39 | 0 | 8 |
| Beech | A | 5.4 | 28.7 | 63/37/0 | 82 | 43 | 39 | 12 | 5 |
| Beech3 | B | 13.9 | 73.9 | 83/17/0 | 45 | 11 | 35 | 9 | 2 |
| Cedar | A | 6.4 | 18.2 | 0/>99/<1 | 64 | 28 | 36 | 0 | 6 |
| Cedar | B | 11.5 | 32.8 | 0/>99/<1 | 41 | 7 | 34 | 0 | 7 |
Table 1. Ethanosolv extraction results. Obtained yields, aromatic distribution and linkages for the different extractions performed on biomass. *Sulfuric acid is used as acid. 1Yield of lignin (wt%)/Lignin content in the feedstock as determined by Klason lignin determination. 2Hemicellulose and S-condensed present in the product. 332% of the S-units are condensed.
| Source | Conditions | Mn (g/mol) | Mw (g/mol) | Ð |
| Walnut | A | 1096 | 1805 | 1.65 |
| Walnut | B | 1174 | 2934 | 2.50 |
| Walnut | C* | 1248 | 2930 | 2.35 |
| Pine | A | 1331 | 3071 | 2.31 |
| Pine | B | 1319 | 3596 | 2.73 |
| Beech | A | 1645 | 3743 | 2.28 |
| Beech | B | 1368 | 4303 | 3.14 |
| Cedar | A | 860 | 1626 | 1.89 |
| Cedar | B | 1188 | 3292 | 2.77 |
Table 2: Molecular weights of the obtained lignins.
| Source | Conditions | Yield (%) | S/G/H ratio | Total β-O-4 | S acetal (wt%) | G acetal (wt%) | H acetal (wt%) | Total acetal yield (wt%) |
| Walnut | A | 5.0 | 45/46/9 | 72 | 4.5 | 5.9 | 2.1 | 12.5 |
| Walnut | B | 15.2 | 59/37/4 | 74 | 3.6 | 4.7 | 1.0 | 9.3 |
| Walnut | C* | 16.2 | 65/33/2 | 45 | 3.8 | 3.9 | 0.6 | 8.3 |
| Pine | A | 3.5 | 0/>99/<1 | 59 | 0 | 9.9 | 0.3 | 10.2 |
| Pine | B | 4.0 | 0/>99/<1 | 46 | 0 | 1.1 | 0 | 1.1 |
| Beech | A | 5.4 | 63/37/0 | 82 | 7.7 | 6.7 | 0 | 14.4 |
| Beech | B | 13.9 | 83/17/0 | 45 | 3.6 | 3.4 | 0 | 7.0 |
| Cedar | A | 6.4 | 0/>99/<1 | 64 | 0 | 8.1 | 0.1 | 8.2 |
| Cedar | B | 11.5 | 0/>99/<1 | 41 | 0 | 4.7 | 0 | 4.7 |
Table 3: Acetal yields of lignin depolymerization. Yields of phenolic 2-arylmethyl-1,3-dioxolones (acetals) obtained from depolymerization of lignin from different sources. Conditions: 50 mg lignin, 60 wt% ethylene glycol, 10 wt% Fe(OTf)3, solvent: 1,4-dioxane, 140 °C (1 mL total volume), 15 minutes (step 7).
| Source | Conditions | Lignin extraction yield (%) | β-O-4 | β'-O-4 | Total β-O-4 | Total acetal yield (wt%) | Overall acetal yield corrected for lignin extraction yield (wt%)1 |
| Walnut | A | 5.0 | 34 | 38 | 72 | 12.5 | 0.63 |
| Walnut | B | 15.2 | 20 | 54 | 74 | 9.3 | 1.41 |
| Walnut | C* | 16.2 | 10 | 35 | 45 | 8.2 | 1.33 |
| Pine | A | 3.5 | 22 | 37 | 59 | 10.2 | 0.36 |
| Pine | B | 4.0 | 7 | 39 | 46 | 1.1 | 0.04 |
| Beech | A | 5.4 | 43 | 39 | 82 | 14.4 | 0.78 |
| Beech | B | 13.9 | 11 | 35 | 45 | 6.9 | 0.96 |
| Cedar | A | 6.4 | 28 | 36 | 64 | 8.2 | 0.52 |
| Cedar | B | 11.5 | 7 | 34 | 41 | 4.7 | 0.54 |
Table 4: Overall acetal yield corrected with extraction yield. Yields of phenolic 2-arylmethyl-1.3-dioxolanes (acetals) obtained from depolymerization of lignin from different sources corrected for lignin extraction yield. 1Calculation: 100*(lignin yield/100)*(total acetal yield/100). Conditions: 50 mg lignin, 60 wt% ethylene glycol, 10 wt% Fe(OTf)3, solvent: 1,4-dioxane, 140 °C, 15 min (reaction via step 6 & work-up via step 7).
