Figure 1 represents an overview of the experimental strategy. The main steps of the protocol and an indicative timeline for the generation of TERRA-MS2 clones in AGS cells are shown (Figure 1A). At day 1, multiple wells of a 6 well plate are transfected with the MS2 cassette and sgRNA/Cas9 expressing vectors (shown in Figure 1B). Two different subtelomere 15q-specific guide RNA sequences are cloned in the Cas9 nickase-expressing pX335 vector, generating two sgRNA-pX335 vectors that are co-transfected with the MS2 cassette. One well of the plate can be transfected with a GFP expressing vector to verify the transfection efficiency. At day 2, the cells are trypsinized and transferred from a single well to a 10 cm dish containing selective medium. At day 3, medium should be changed as most untransfected cells will be dead. Cells are grown in selection until clones are visible and ready to be picked. During this time, cells should not be trypsinized and culturing medium should be changed every 1-2 days for the first week and then every 2-3 days afterwards. Once single clones are visible, they are picked and transferred in a 96 well plate where they are allowed to grow until reaching 80-90% of confluence. At this point, clones are split in 4 different 96 well plates, which are marked in Figure 1 with a color code. Once the clones cultured in the DNA plate (green) reach 80% confluence, they are lysed and genomic DNA extracted for PCR screening; the clones in the backup plate (blue) will be kept in culture until the results of the PCR screening, while the red freezing plates are frozen at -80°C. Figure 2B shows representative results of a PCR screening of neomycin-resistant clones. For this screening, two sets of primers are used: one primer pair (MS2 primers) annealing within the neomycin gene and subtelomere 15q sequence is used to verify the presence of the MS2 cassette (a representative image of the primers localization is shown in Figure 2A). A second primer pair (CTR primers) annealing at an internal chromosomal region is used to verify the presence of false negative clones (primer sequences are indicated in Table 1). Negative clones for the integration of the cassette should be negative to the MS2 primers' amplification and positive to the CTR primers' amplification. Technical problems in genomic DNA extraction resulting in the absence of genomic DNA or its contamination would preclude PCR amplification from MS2 and CTR primer reactions. In these cases, the clones involved can be re-screened by PCR upon extraction of genomic DNA from the backup plate. Bands obtained from MS2 primers amplification of positive clones can be gel-extracted and sequenced using the MS2 primers, in order to confirm the presence of ten MS2 sequences.
The clones that are positive at PCR screening are cultured from the 96 well backup plate (the blue plate in Figure 1) to a 6 well plate, then lysed for genomic DNA extraction and Southern blot analyses. Figure 2D shows representative images of a gel (left) and membrane hybridized with a radioactively labelled MS2 sequence specific probe (right) of a Southern blot screening of PCR positive clones. For confirmation of the MS2 sequences integration at subtelomere 15q, genomic DNA extracted from each clone should be digested with two different restriction enzymes (NcoI and BamHI) in two separate digestion reactions. The NcoI and BamHI restriction enzymes are suitable for verification of the MS2 cassette integration at subtelomere 15q. Other enzymes can also be used. The gel shows a complete digestion of genomic DNA using the BamHI restriction enzyme, as indicated by the presence of a smear. The results from Southern blot indicate that one clone is positive for the integration of the MS2 cassette at subtelomere 15q while several clones show multiple integration events of the cassette, as indicated by the presence of multiple bands. A positive clone should be analyzed by a second Southern blot upon NcoI digestion of genomic DNA21. A representative image of a subtelomere 15q containing the MS2 cassette and the position of BamHI and NcoI restriction enzyme sites is shown in Figure 2C.
The clones positive to PCR and Southern blot analyses are infected with a Cre-GFP expressing adenovirus in order to remove the neomycin gene present in the MS2 cassette. Figure 3A depicts a MS2-tagged subtelomere 15q before and after Cre expression. An image of a clone positive for the MS2 cassette integration at subtelomere 15q infected with a Cre-GFP expressing adenovirus is shown in Figure 3B. For a complete removal of the neomycin gene in all cells, the infection efficiency should reach approximately 100%. As shown in Figure 3C, in order to verify the elimination of the neomycin gene, Cre-GFP infected cells are split in three plates after 48 hours from the infection. One plate will be cultured in the presence of neomycin. All cells in this plate should die within 6-7-days. In a second plate, cells will be allowed to grow in complete medium without selection up to 80-90% confluence. At this point, genomic DNA is extracted and analyzed by Southern blot. The third plate is cultured in complete medium to allow the preparation of frozen stocks of the clone and for RNA extraction and RT-qPCR analyses of TERRA-MS2 transcript expression. Figure 3D shows representative images of Southern blot analyses of a positive clone before and after Cre-GFP infection. DNA agarose gel (image on the left) confirms the complete digestion of the genomic DNA using the NcoI restriction enzyme. Southern blot analysis was performed using a radioactively-labelled MS2-sequence specific probe (image on the right). This analysis confirms the removal of the neomycin gene. The presence of a smear in the Cre-GFP infected sample confirms the telomeric integration of the MS2 sequences. The position of the NcoI restriction sites within the subtelomere 15q is shown in Figure 3A.
Once the elimination of the neomycin resistance gene is confirmed, the clones can be tested for the expression of TERRA-MS2 transcripts. To this aim, total RNA is extracted from each clone and run on a denaturating MOPS gel to confirm its integrity (Figure 4A). Ribosomal RNA bands should be visible at 4,700 bases (rRNA 28S) and 1,900 bases (rRNA 18S). Retrotranscription reaction is performed using a telomeric repeat-specific primer and reference gene-specific primer (Figure 4B, top) while qPCR analyses of TERRA expression are performed using two sets of primers: one primer pair annealing within the MS2 sequence and the subtelomere 15q sequence; a second pair of primers annealing within the subtelomere 15q. Primer sequences are indicated in Table 1. The graph presented in Figure 4B shows RT-qPCR analyses of TERRA expression from AGS WT cells and two different TERRA-MS2 clones. These analyses confirm the expression of TERRA-MS2 transcripts in the two clones at levels that are comparable to TERRA transcripts expressed from subtelomere 15q in WT cells. These data indicate that the two TERRA-MS2 clones selected are suitable for the analyses of TERRA-MS2 transcripts by live cell imaging.
In order to visualize TERRA-MS2 transcripts in living cells, the selected clones are infected with a retrovirus expressing the MS2-GFP fusion protein. Figure 5A shows the procedure to generate MS2-GFP expressing retrovirus, as described in protocol step 4. AGS WT cells and TERRA-MS2 clones are infected in glass-bottomed dishes. After 24 h from the infection, cells are analyzed by fluorescence microscopy. The specificity of the signal is confirmed by the presence of TERRA-MS2-GFP foci detected in the nucleus of TERRA-MS2 clones and not in AGS WT cells (Figure 5B). In a population of MS2-GFP expressing cells, heterogeneity in terms of MS2-GFP levels among cells is expected and cells expressing low levels should be chosen for the imaging analyses. Alternatively, the MS2-GFP expressing cells can be sorted by FACS prior microscopy analyses in order to collect the subpopulation of cells expressing low levels of GFP. We have previously detected TERRA-MS2-GFP foci in 40% to 60% of cells of AGS TERRA-MS2 clones21. In these cells, one to four TERRA-MS2-GFP foci were imaged per cell. TERRA-MS2-GFP foci showing distinct sizes and dynamics can be detected21. TERRA-MS2 clones can be used to study the dynamics of single-telomere TERRA transcripts in living cells. As an example of this application, Figure 5C shows representative images from microscopy analyses of a TERRA-MS2 clone expressing the MS2-GFP fusion protein and the telomere-binding protein TRF2 fused to mCherry in order to visualize MS2-tagged TERRA transcripts and telomeres in living cells. Using this approach, we have previously observed that TERRA-MS2-GFP foci co-localize with telomeres in 44% of the cells, indicating that TERRA transcripts only transiently co-localize with chromosome ends in AGS cells21.
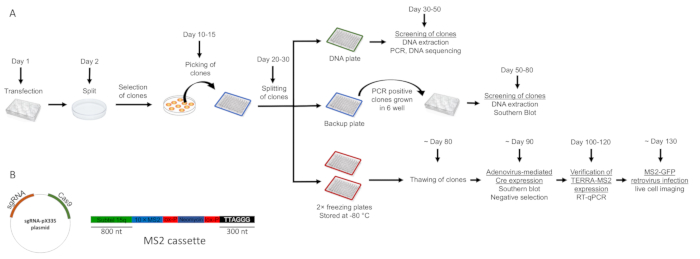
Figure 1. Overview of the experimental strategy and indicative timeline for the generation of TERRA-MS2 clones in AGS cells. A) The steps described in the protocol and an indicative time line for the selection of TERRA-MS2 clones are shown. Cells are transfected in a 6 well plate with the linearized MS2 cassette and sgRNA/Cas9 expressing vectors. A color code is used to distinguish the DNA plate, the backup plate and the freezing plates indicated in the protocol section. B) The MS2 cassette consists of an 800 nt long subtelomere 15q sequence followed by 10 repetitions of the MS2 sequences and a neomycin resistance gene flanked by lox-p sites and terminates with a 300 nt long telomeric repeat tract at its 3' end. sgRNA/Cas9 expressing vectors were generated by cloning subtelomere 15q-specific guide RNA sequences in pX335 vector using the BbsI restriction site21,22. Two different sgRNA-pX335 vectors were generated and a 1:1 mix of the two vectors was used for the transfection. The sequences of the sgRNAs are indicated in Table 1. Please click here to view a larger version of this figure.
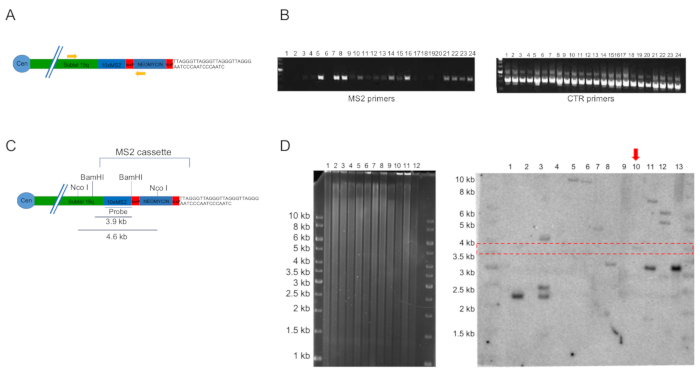
Figure 2. Representative images of PCR and Southern blot screening of neomycin resistant clones.A) Representative image of subtelomere 15q containing the MS2 cassette. The position of the MS2 primers (MS2-subtel15q-primer-S and MS2 primer AS) used for PCR screening is indicated. The loxP sites present within the cassette are shown in red. B) Representative image of PCR screening of neomycin resistant clones using two set of primers, MS2 primers and CTR primers (CTR prime S and CTR primer AS). C) Representative image of subtelomere 15q containing the MS2 cassette. BamHI and NcoI restriction sites and the MS2 probe used for the Southern blot screening are shown. D) Left: 10 µg of genomic DNA per clone were digested with BamHI and run on an agarose gel. The image was acquired after an overnight run at 30 volts. Right: genomic DNA digested with BamHI restriction enzyme was transferred to a positively charged nylon membrane and hybridized with a radioactively-labelled MS2 sequence-specific probe. The red box indicates the expected size of the positive band. The red arrow indicates one positive clone. Please click here to view a larger version of this figure.
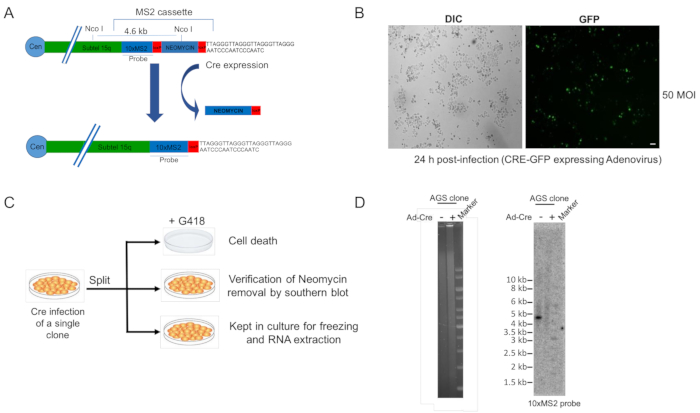
Figure 3. Elimination of the neomycin resistance gene and validation experiments. A) Representative image of subtelomere 15q containing the MS2 cassette before and after Cre expression. The MS2 specific probe used for Southern blot analyses and NcoI restriction enzyme sites are shown. The loxP sites present within the cassette are shown in red. B) Fluorescence microscopy analysis of a TERRA-MS2 clone infected with Cre-GFP expressing adenovirus. Representative image acquired at a single focal plane is shown. MOI, multiplicity of infection, is the ratio between the number of viruses used for the infection and the number of host cells. Scale bar: 30 µm C) Overview of the experimental strategy used to verify the elimination of the neomycin gene. Cre-GFP infected cells are split in three plates after 48 hours from the infection for i) negative selection, ii) Southern blot analyses and iii) preparation of frozen cell stocks and RNA extraction. D) Southern blot analyses of a TERRA-MS2 clone before and after Cre-GFP adenovirus infection. 10 µg of genomic DNA were digested with the restriction enzyme NcoI. Agarose gel confirms that complete digestion is achieved in both clones (left). The digested genomic DNA was transferred to a positively charged nylon membrane and hybridized using a MS2 sequence-specific probe. Complete elimination of the neomycin gene is confirmed by the absence of the specific band. Please click here to view a larger version of this figure.
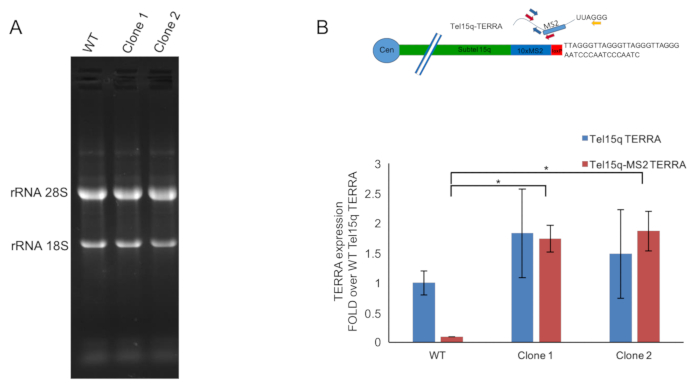
Figure 4. RT-qPCR analyses of Tel15q-TERRA and Tel15q-TERRA-MS2 transcripts expression. A) Representative image of a MOPS gel showing total RNA extracted from AGS WT cells and TERRA-MS2 clones. Bands corresponding to the ribosomal RNAs 28S and 18S are indicated. B) Top: A schematic of the MS2-tagged subtelomere 15q expressing TERRA transcripts. Primers used for TERRA retrotranscription (yellow) and qPCR analyses of Tel15q-TERRA (blue) and Tel15q-MS2 TERRA (red) are shown. The loxP site is shown in red. Bottom: RT-qPCR analyses of Tel15q-TERRA and Tel15q-MS2 TERRA transcripts expression in AGS WT cells and TERRA-MS2 clones. *p < 0.05, unpaired t-test. Please click here to view a larger version of this figure.
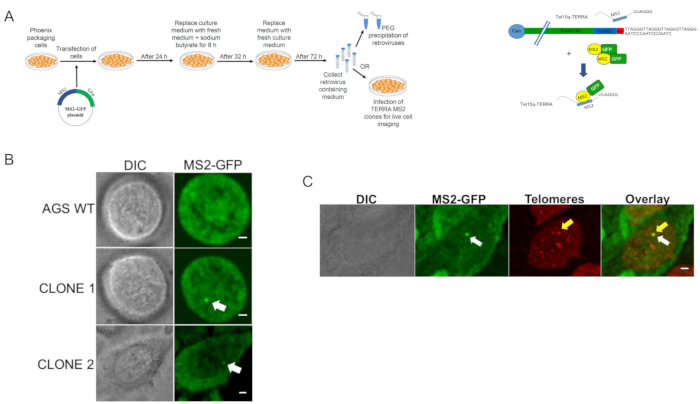
Figure 5. Live cell imaging analyses of TERRA-MS2 clones. A) An overview of the procedure to produce a MS2-GFP expressing retrovirus, as described in protocol step 4. A schematic of the MS2-tagged subtelomere 15q and TERRA transcripts recognized by the MS2-GFP fusion protein is shown on the right. B) Representative fluorescence microscopy image of AGS WT and two TERRA-MS2 clones expressing MS2-GFP. TERRA-MS2-GFP foci are indicated by arrows. Images were acquired using a spinning disc confocal microscope equipped with an EMCCD camera. The images were captured using a 100X/1.46 apochromat objective in an imaging chamber maintained at 37 °C with 5% CO2. A 488 laser was used as light source. Scale bar: 5 µm. C) Live cell imaging analyses of TERRA-MS2-GFP foci and TRF2-mCherry labeled telomeres. A co-localization event between a TERRA-MS2-GFP focus and a single telomere is shown. Images were acquired as in B, using 488 and 520 lasers as light source. A maximal projection of a z stack experiment performed at a single time point of time-lapse imaging experiment is shown. Scale bar: 5 µm. Please click here to view a larger version of this figure.
| Primer name | Primer sequence | Application |
| MS2-subtel15q-primer-S | TGCATTAAAGGGTCCAGTTG | MS2 primer forward used for PCR screening of neomycin resistant clones |
| MS2 primer AS | CCTAACTGACACACATTCCACAGA | MS2 primer reverse used for PCR screening of neomycin resistant clones |
| CTR primer S | TGT ACG CCA ACA CAG TGC TG | CTR primer forward used in PCR screening of neomycin resistant clones |
| CTR primer AS | GCT GGA AGG TGG ACA GCG A | CTR primer reverse used in PCR screening of neomycin resistant clones |
| Tel15q-S1 | GCAGCGAGATTCTCCCAAGC | Sense primer used to detect Tel15q and Tel15q-MS2 TERRA expression by qPCR |
| hTel15q-AS | TAACCACATGAGCAATGTGGGTG | Antisense primer used to detect Tel15q and Tel15q-MS2 TERRA expression by qPCR |
| Tel15q-MS2-AS | ATGTTTCTGCATCGAAGGCATTAGG | Antisense primer used to detect Tel15q-MS2 TERRA expression by qPCR |
| TERRA-RT-primer | CCCTAACCCTAACCCTAACCCTAACCCTAA | Primer used for retrotranscription of TERRA transcripts |
| sgRNA1 | TTGGGAGAATCTCGCTGGCC | Sequence of the short guide RNA 1 cloned in pX335 vector and used to direct Cas9 nickase enzymatic activity to subtelomere 15q |
| sgRNA2 | TGCATTAAAGGGTCCAGTTG | Sequence of the short guide RNA 2 cloned in pX335 vector and used to direct Cas9 nickase enzymatic activity to subtelomere 15q |
Table 1: List of the primers used in this study. A list of the primers and the sequences of the sgRNAs used in this protocol are provided. Sequences are 5' to 3'.
