Evaluation of Host-Pathogen Responses and Vaccine Efficacy in Mice
Summary
Here we present an elegant protocol for in vivo evaluation of vaccine effectiveness and host immune responses. This protocol can be adapted for vaccine models that study viral, bacterial, or parasitic pathogens.
Abstract
Vaccines are a 20th century medical marvel. They have dramatically reduced the morbidity and mortality caused by infectious diseases and contributed to a striking increase in life expectancy around the globe. Nonetheless, determining vaccine efficacy remains a challenge. Emerging evidence suggests that the current acellular vaccine (aPV) for Bordetella pertussis (B. pertussis) induces suboptimal immunity. Therefore, a major challenge is designing a next-generation vaccine that induces protective immunity without the adverse side effects of a whole-cell vaccine (wPV). Here we describe a protocol that we used to test the efficacy of a promising, novel adjuvant that skews immune responses to a protective Th1/Th17 phenotype and promotes a better clearance of a B. pertussis challenge from the murine respiratory tract. This article describes the protocol for mouse immunization, bacterial inoculation, tissue harvesting, and analysis of immune responses. Using this method, within our model, we have successfully elucidated crucial mechanisms elicited by a promising, next-generation acellular pertussis vaccine. This method can be applied to any infectious disease model in order to determine vaccine efficacy.
Introduction
Vaccines represent one of the greatest public health achievements of the 20th century, yet we still do not fully understand the mechanisms by which successful vaccines stimulate protective immunity. The identification of molecular signatures (e.g., cell activation markers, expansion of cellular subtypes, and patterns of gene expression) induced after vaccination provides a plethora of information for predicting and generating an efficacious immune response. The complexity of host-pathogen responses cannot be adequately replicated using in vitro cell culture systems1. In vivo vaccine models are designed to concomitantly evaluate multiple immune cell types within the host. This provides an advantage when characterizing vaccine antigen processing and presentation, differential cytokine secretion, and expansion of immune cells. The protocol described here provides a detailed method to determine vaccine efficacy through evaluation of the systemic and local immune responses and quantification of pathogen burden in tissues of interest. The example provided here tests the efficacy of an experimental vaccine for the pathogen Bordetella pertussis (B. pertussis).
B. pertussis is a gram-negative bacterium that is the etiological agent of the respiratory disease whooping cough (pertussis)2,3. Close contact with infected individuals (symptomatic or asymptomatic) leads to transmission, colonization, and disease. Despite significant global vaccine coverage4, pertussis is considered a resurging disease in many nations around the world and is a major cause of preventable childhood deaths5,6,7,8. In 2015, B. pertussis and pertussis were included in the National Institute of Allergy and infectious Diseases (NIAID) emerging infectious pathogen/disease list, emphasizing the need for development of a better vaccine that confers long-lived protective immunity.
Currently, an active area of investigation to control pertussis resurgence is development of a next-generation acellular pertussis vaccine (aPV) with an optimal combination of novel adjuvants and antigens to mimic the immune response elicited by the whole-cell pertussis vaccine (wPV)9. Using the protocol described, we recently reported that the modification of a current FDA-approved aPV by the addition of a novel adjuvant, Bordetella colonization factor A (BcfA), resulted in more efficient reduction of B. pertussis bacterial load from mouse lungs10,11. This increased protection was accompanied by the skewing of an alum-induced Th1/Th2 immune response to the more protective Th1/Th17 immune profile10. This protocol is detailed and comprehensive, enabling the investigator to obtain maximal information through concurrent evaluation of host and immune responses to a variety of pathogens.
The protocol described here follows the representative vaccine schedule, shown in Figure 1, to ensure optimal host immune responses.
Protocol
All experiments with live animals were conducted following a protocol approved by The Ohio State University IACUC in accordance with IACUC guidelines. C57BL/6 mice were used in all immunizations and infections. Both male and female mice are used in each group as per NIH guidelines. The number of animals per group was determined by power calculations based on the predicted differences in outcome among experimental groups. For example, 8 mice per group will yield 80% power at α = 0.05 (2-sided) for a 2-sample t-test to detect differences in the outcome of interest of 1.33 standard deviations (SDs).
1. Immunization of Mice
- Prepare a master vaccine mix in a sterile physiological buffer such as saline or DPS.
NOTE: The total volume in the mix should include 10% more than that required for immunization of the whole group. Concentrations of the vaccine composition are detailed below in steps 1.1.1 and 1.1.2. Transport the items to the vivarium on ice.- For B. pertussis studies, include the following vaccine groups: aPV and aPV + BcfA. In addition, use alum, vaccine antigens alone (FHA, and Prn), and naïve (non-immunized) mice as control groups.
NOTE: aPV is 1/5th the human dose of an FDA-approved aPV, which is composed of tetanus toxoid, reduced diphtheria toxoid, pertussis toxoid (PT), filamentous hemagglutinin (FHA), and pertactin (Prn) adsorbed to aluminum salts. One human dose of current pertussis aPV is 0.5 mL, therefore, 1/5th of a dose is 0.1 mL. Volumes for aPV + BcfA are 0.1 mL of the aPV (1/5th the human dose) + 0.046 mL of BcfA (30 µg) per mouse. The maximum volume that can be safely delivered to one muscle is 0.1 mL. Because the aPV + BcfA vaccine volume is greater than 0.1 mL, the vaccine must be delivered into both shoulders, divided roughly equally, in order to be administered safely. - To prepare an experimental vaccine, for one dose combine 1 µg of FHA and 0.5 µg of Prn with 50 µg of aluminum hydroxide gel in sterile PBS. Allow the experimental aPV to mix on a laboratory roller for 15 min at room temperature (RT). Afterwards, spin the tube at 18,000 x g for 5 min at 4 °C. Remove the supernatant and resuspend the contents in 1 mL of sterile PBS.
- For B. pertussis studies, include the following vaccine groups: aPV and aPV + BcfA. In addition, use alum, vaccine antigens alone (FHA, and Prn), and naïve (non-immunized) mice as control groups.
- In the vivarium, set up the anesthesia machine (Figure 2).
NOTE: Ensure there are adequate levels of oxygen and isoflurane in their respective tanks before use. Weigh the isoflurane scavenger canister and replace when its weight increases by 50 g. - Thoroughly clean the biological safety cabinet (BSC) and place the clean induction chamber inside the BSC. Connect the anesthesia and scavenger lines from the anesthesia machine to the induction chamber. Open the oxygen tank via the regulator to ensure oxygen is flowing and set the level to 1.5-2 L/min.
- Place mice of desired age (6-12 weeks) into the induction chamber. Turn on the isoflurane to 2.5-3% in order to lightly anesthetize the mice. Monitor the animals until they are not moving in the chamber and their breathing has slowed.
- Test the level of induction by toe-pinching, observing for any reflexive movement. If there is none, then the mice are ready to inject.
NOTE: In this experiment, the whole cage of animals were anesthetized at once. One can choose to anesthetize animals individually for injection or to inject the animals without anesthesia. Either of these methods is appropriate. - Meanwhile, prepare 1 mL insulin syringes (28G1/2) with 0.1 mL of vaccine each. When animals are fully induced, remove one animal from the chamber and lay the mouse ventrally on a towel or bench pad.
- Administer the vaccine intramuscularly. With the syringe at a 45° angle and with the bevel facing up, insert the syringe ~5 mm into the deltoid of the mouse and inject the vaccine.
NOTE: The hind quarter/flank may be used as an injection site instead of the deltoid. - After injection, keep the needle inserted in the muscle for ~5-10 s. Once the volume is delivered, rotate the syringe at 180° so that the bevel faces away to create a seal and prevent the leakage of the vaccine. Then, slowly pull the needle out of the injection site.
- Return the mouse to the cage. Repeat the procedure with all animals in the designated group.
- Turn off the isoflurane and flush the induction chamber with oxygen before anesthetizing the next cage of mice.
- Monitor the animals until they are all alert and moving in the cage. Return the cage to the housing location.
- Monitor animals every 12 h post vaccination, up to 48 h, for clinical symptoms of morbidity such as rough/unkempt coat, slow gait, or labored breathing. If symptoms persist, confer with the institutional veterinarian and remove the mice from the study if needed.
- Four weeks after initial immunization, boost mice by anesthetizing and administering intramuscular injections into the right deltoid of the mouse with the same vaccine formulation that was previously given.
- Again, monitor animal for any clinical symptoms. House animals for an additional 2 weeks and then proceed with infection.
2. Growth of B. pertussis Strains and Preparation of Infection Inoculum
- From frozen stocks [cryopreserved at -80 °C in 15% glycerol, frozen from planktonic growth in Stainer-Scholte (SS) media12], streak out B. pertussis on Bordet-Gengou (BG) agar13 plates supplemented with 10% defibrinated sheep's blood and 100 µg/mL streptomycin (10% BG + Sm) for selection. Incubate the plate at 37 °C for 4 days.
- Pick ~8-10 individual colonies of the strain(s) from the plate and inoculate 3 mL of SS media supplemented with 22.8 mM L-cysteine, 3.6 mM ferrous sulfate, 3.3 mM niacin, 48.8 mM glutathione, 227 mM ascorbic acid, 1 mg/mL heptakis, and 100 µg/mL streptomycin. Grow the culture at 37 °C in a roller drum at 60 rpm for 16-24 h.
- The next day, dilute the primary culture 1:600, 1:300, 1:120, 1:60, 1:30, and 1:15 in 3 mL of supplemented SS media. Incubate the culture at 37 °C in roller drum at 60 rpm for 16-24 h.
- Perform optical density measurements at 600 nm (OD600) for the bacterial cultures. Using spectrophotometer cuvettes, dilute the cultures 1:10 by adding 0.1 mL of the liquid cultures to 0.9 mL of sterile PBS.
- Select a culture with OD600≈ 0.8-1.2. Calculate the volume needed to obtain 1 OD (Figure 3). Spin down the volume in a 1.5 mL microcentrifuge tube at 6000 x g for 5 min at 25 °C. Aspirate the supernatant and resuspend bacterial cell pellet in 1 mL of sterile PBS to obtain a density of 1 OD/mL [1-3 x 109 colony forming units (CFU)/mL].
- To prepare an inoculum of ~5 x 105 to 1.5 x 106 CFUs/mouse in 40-50 µL, vortex and serially dilute the bacterial solution (from step 2.5) 100-fold in 1 mL of sterile PBS to achieve ~1-3 x 107 CFUs/mL (Figure 4A). Transport the inoculum to the vivarium on ice.
3. Murine Intranasal Infection Model
- In the vivarium, set oxygen and anesthesia conditions as described in steps 1.3 and 1.4. As described in step 1.5, place the mice in the chamber to anesthetize. Monitor the animals until anesthesia takes effect.
- When the mice are anesthetized, remove one animal at a time from the induction chamber. Pick up the mouse by the scruff of the dorsal thorax, behind the shoulder blades and neck. Position the mouse upright to access the nose.
- Using a 200 µL pipette tip, slowly apply 40-50 µL of the bacterial inoculum to the nares of the mouse (20-25 µL in each nare). Hold the mouse until the entire inoculum has been inhaled by the animal. If some of the inoculum bubbles out, collect the bubbles and re-instill into the nares. Deliver an equal amount of sterile PBS to one group of mice as negative control.
- As done in steps 1.10-1.12, return the inoculated animals to their cage and monitor the animals until they are alert and moving. Return the cage back to its original housing space on the rack. Flush the induction chamber with oxygen to remove the residual isoflurane before anesthetizing the next set of animals. Monitor the animals for 48 h post-infection.
- In the laboratory, plate serial dilutions of the inoculum, in sterile PBS, to verify the actual CFUs delivered to the mice. Plate 0.1 mL of dilutions on 10% BG + Sm plates (Figure 4A). Place the plates in the 37 °C incubator and grow for 4 days. Count and calculate the CFUs from the inoculum delivered to the mouse (Figure 4B).
4. Harvesting of Animal Tissue after Infection
- Prepare for mouse dissection and tissue harvest.
- Sterilize all tools (coarse and fine scissors, curved scissors, fine forceps, pellet pestles, and triangle spreaders) needed for processing tissues in an autoclave. Seal the tools in sterilization pouches. Wrap the glass dounce homogenizers in foil and add autoclave tape to indicate sterility.
- Prepare agar plates 1 or 2 days prior to the tissue harvest to ensure that plates are solidified prior to use. For B. pertussis, use 10% BG + Sm. Label plates for nasal septum, trachea, and lung for each mouse with the desired dilutions, determined empirically for each experiment.
- Fill and label CFU dilution tubes. Add 0.9 mL of sterile PBS to sterile 1.5 mL microcentrifuge tubes based the on number of organs to process and dilutions per organ.
- Fill and label tubes for tissue collection:
- Nasal septum and trachea: fill a sterile 1.5 mL microcentrifuge tube with 0.3 mL of sterile 1% casein in PBS (Ca2+ and Mg2+-free). Store at 4 °C.
- Lungs: add 2 mL of sterile 1% casein in PBS to a sterile 15 mL conical tube and store at 4 °C. To harvest lungs for histology, add 2 mL of 10% neutral buffered formalin (NBF) and store at RT.
- Spleen: add 3 mL of RPMI + 5% FBS to a 15 mL conical tube. Store at 4 °C.
- Blood: label two sets of sterile 1.5 mL microcentrifuge tubes, one for whole blood and one for serum.
- Fresh 70% ethanol should be prepared to fill beakers and spray bottles for dissection.
- Transport all materials to the vivarium on a cart on the day of harvest. Clean the BSC and set up the anesthesia machine as in steps 1.3 and 1.4.
- Place the mice to be dissected in the induction chamber and, with oxygen flowing, turn on the isoflurane to 5% and wait until animals are unconscious. Remove mice one at a time, and cervically dislocate the animal as a secondary method to ensure death. Following cervical dislocation, euthanasia should be confirmed by the absence of a respiratory rate and/or absence of the withdrawal reflex (toe pinch test).
- Position the mouse, ventral side facing up, on the dissection board and pin the arms and legs open. Spray down the body of the mouse with 70% ethanol.
- Using forceps, clasp the skin below the abdomen, in the center of the pelvis. Using sharp scissors, make a vertical cut up to the mandible. Then, dissect the skin away from the peritoneal wall by pushing the two layers apart, using small motions with the scissors closed. Place the skin to the sides of the carcass to create an unobstructed field for sterile organ harvest.
- Grasp the intact peritoneum below the rib cage at or around the liver and cut it open moving towards the rib cage. Expose the lower digestive tract and move the colon to the left revealing the spleen. Using curved forceps, grasp the spleen and dissect away the connective tissue with scissors. Place the spleen in a 15 mL conical tube containing 3 mL of RPMI + 5% FBS and place the tube on ice.
- Use forceps to stabilize the rib cage at the xiphoid process and cut open the diaphragm. Lungs should deflate and fall towards the spine. Cut open the rib cage at the sides to remove the breast plate.
- Isolate and remove the superior lobe of the right lung14, cutting the pulmonary arteries and veins. Place the lobe in a 15 mL conical tube containing 10% NBF and place the tube at RT for at least 24 h to fix the tissue. Isolate the remaining lobes of the right lung and the left lung. Place lobes in a 15 mL conical tube containing 2 mL of 1% casein in PBS and place it on ice.
- After cutting the pulmonary veins and arteries to dissect out the lungs, allow the chest cavity to fill with blood as the mouse exsanguinates. Using a 1 mL pipetman with tip, collect the blood and transfer it to the pre-labeled 1.5 mL microcentrifuge tube. Place the tube on ice.
- Then, from the neck, remove the soft tissue (parotid, sublingual, and submaxillary glands and lymph nodes) that covers the trachea. Open and remove the protective membranes surrounding the trachea. Delicately, using fine forceps and scissors, separate the trachea from the esophagus and any other connective tissue from top of the collarbones to bottom of the mandible.
- Using forceps, hold on to the trachea and cut the trachea at the top of the clavicle. Pull the trachea down to maximize elasticity and cut the trachea at the bottom of the mandible right above the larynx, isolating as much as possible (~1 cm). Place the organ in a pre-labeled 1.5 mL microcentrifuge tube with 0.3 mL of 1% casein in PBS and place it on ice.
- Unpin the mouse and lay it ventrally, with the mouse facing the researcher for clear access to the nose. Spray the head of the mouse with 70% ethanol and, using hands, grasp the mouse directly behind the skull.
- Using scissors, cut away soft flesh of the snout, starting from the bottom of the nose and moving upward. In addition, remove the fur, skin, and whiskers around the nose. Then, with forceps and curved scissors, insert the tips of scissors into nares with the curves pointed outward, and cut open the nasal passage towards the eyes, creating a V-like formation.
- Using fine forceps, collect the nasal septum and soft tissue within the created formation. Place the tissue in a pre-labeled 1.5 mL microcentrifuge tube with 0.3 mL of 1% casein in PBS and place it on ice.
- Dispose of the carcass in a biohazard bag. Before dissecting the next animal, rinse the tools with deionized water, and then place in a beaker filled with 70% ethanol. Remove the tools, shake off excess ethanol, and then proceed with the next dissection
- Dissect each mouse following steps 4.3-4.14.
- Process the tissues as described below.
5. Processing of Spleen
- To dissociate spleens, place a 70 µm cell strainer in a 60 mm tissue culture dish. Pour the spleen and accompanying media into the filter, using the plunger of a 3 mL syringe. Carefully mash the organ until it is completely dissociated and filtered through cell strainer.
- Rinse the filter with 3 mL of RPMI + 5% FBS into the tissue culture dish. Collect the cell suspension and place it in its original 15 mL tube. Centrifuge the tubes at 450 x g for 5 min at 4 °C to pellet the cells.
- Aspirate or decant the supernatant and lyse the red blood cells by resuspending the pellet in 2 mL of ammonium chloride potassium (ACK) buffer (150 mM ammonium chloride, 10 mM potassium bicarbonate, 0.1 mM sodium EDTA). Incubate for 3 min at RT (25 °C).
- Fill the tubes up to 8 mL with RPMI + 5% FBS and centrifuge tubes at 450 x g for 5 min at 4°C. Aspirate or decant the supernatant. Resuspend the cell pellets in 5 mL of RPMI + 5% FBS and count the splenocytes using a hemocytometer and microscope.
- Calculate the volume of cells needed to stimulate 2.5 x 106 cells per well. In a 48-well plate, seed the cells in 0.5 mL of T-cell media (RPMI 1640 + 10% FBS, 10 µg/mL gentamicin, and 50 µM β-mercaptoethanol) per well.
- Based on the above calculation, place the required volume of the cell suspension into a fresh tube and pellet-cells at 450 x g for 5 min at 4 °C.
- Resuspend the pellet in the required volume of T-cell media. For a B. pertussis infection model, test the antigen-specific cytokine response to vaccine antigens: pertactin (Prn) and filamentous hemagglutinin adhesion (FHA). Therefore, 3 treatments are needed: 1) no stimulation (NS, as negative control), 2) Prn, and 3) FHA. For the assay, 7.5 x 106 cells in 1.5 mL of T-cell media is required (2.5 x 106 cells per 0.5 mL).
- Aliquot 0.5 mL of the cell suspension into a 48-well plate.
- To stimulate cells, mix antigens at 2 times the desired concentration in T-cell media. Final concentrations of Prn and FHA = 1 µg/mL and 0.5 µg/mL, respectively. Therefore, mix 2 µg/mL Prn or 1 µg/mL FHA in 0.5 mL. Then, aliquot 0.5 mL of the stimulation medium into each designated well, diluting the antigen treatment to 1x in a final volume of 1 mL in each well.
- Incubate the plates at 37 °C in 5% CO2. Collect supernatants on day 7 and aliquot 0.5 mL of the supernatants into pre-labeled microcentrifuge tubes. Store the samples in a -20 °C until ready to test antigen-specific cytokines by ELISA (Figure 6).
6. Processing of Lungs
- Transfer the lungs (in 2 mL of 1% casein in PBS) into a sterile, 15 mL dounce homogenizer. Use pestle to homogenize tissue. Dissociate until no large particles of tissue remain. Place the homogenized suspension back in the original 15 mL tube.
- Remove a 0.3 mL aliquot for plating CFUs and centrifuge the homogenate at 450 x g for 5 min at 4 °C. Collect and aliquot the supernatant in 0.5 mL aliquots into pre-labeled microcentrifuge tubes. Store the samples in a -20 °C until the analysis of cytokines by ELISA.
- Prepare chosen dilutions by serially diluting 0.1 mL of the homogenized lung suspension in dilution tubes (0.9 mL of sterile PBS). Plate 0.1 mL of empirically chosen dilutions onto pre-labeled 10% BG + Sm plates and spread using sterile triangle spreader.
- Incubate the plates at 37 °C in room air for 4 days.
- Count and calculate CFUs/lung (Figure 6). Briefly, multiply recovered CFU numbers by the dilution factor of the plate, then also by the total volume in which the organ was processed. The CFU calculations are then transformed into Log10 values for each animal and graphed.
- Plates with 30-300 colonies are easily and accurately counted. Plates with less than 6 colonies should not be used for calculations since such low colony numbers introduce error15. To obtain a lower limit of detection, the total volume in which an organ was homogenized is divided by the volume used to spot onto a plate from the undiluted homogenate (e.g., 2 mL total volume divided by 0.1 mL of undiluted homogenate equals the lower limit of 20 CFUs detectable).
7. Processing of Nasal Septum and Trachea
- Homogenize the tissue in the 0.3 mL of 1% PBS/casein with pellet pestle cordless motor homogenizers. Homogenize the tissue for 0.5-1 min until the tissue is largely dissociated. Bacteria should be shed into the suspension.
- Prepare chosen 10x dilutions by serial dilution of 0.1 mL of the homogenized suspension in dilutions tubes containing 0.9 mL of sterile PBS. Dot 0.1 mL of each dilution onto pre-labeled 10% BG + Sm plates and spread the suspension using a triangle spreader that has been sterilized by a 70% ethanol wash and flame.
- Incubate the plates at 37 °C in room air for 4 days.
- Count and calculate CFUs/organ as in step 6.5 (Figure 6). To obtain a lower limit of detection, the total volume in which an organ was homogenized is divided by the volume used to dot on a plate from the undiluted homogenate (e.g., 0.3 mL total volume divided by 0.1 mL of undiluted equals the lower limit of 3 CFUs detectable).
8. Processing of Blood
- Centrifuge the tubes containing isolated whole blood at 18,000 x g for 30 min at 4 °C to pellet red blood cells.
- Carefully take off the serum supernatant and pipet into a pre-labeled serum microcentrifuge tube. Store the tubes in -20 °C until ready for further downstream analysis (i.e., total and specific Ig ELISA).
Representative Results
The model described shows a method to evaluate vaccine efficiency and immune responses during host-pathogen interactions. Figure 1 depicts the representative vaccine schedule used to immunize and infect mice and harvest tissues for analysis. Figure 2 demonstrates the setup of the anesthesia system employed to induce mice, enabling investigators to deliver immunizations and bacterial inoculums. Figure 3 shows example OD600 measurements and calculations to achieve a 1 OD bacterial suspension to prepare 100-fold diluted bacterial inoculum to be delivered to mice. Figure 4 depicts the inoculum preparation used for the infection, the plating scheme of the serial dilutions, and the example calculations used to enumerate CFUs delivered to mice based upon plating serial dilutions. Figure 5 shows antigen-specific recall responses elicited after seven days of stimulation with the vaccine antigen FHA. Immunize spleen cells from the combination vaccine group, Alum/FHA/BcfA, produced IFNγ and IL-17, while significantly downregulating IL-5. This effect promotes a Th1/Th17 polarization of the immune response during B. pertussis infection. Background levels of cytokines were produced by incubation of immunized spleen cells with media alone, indicating that the cytokines detected were recall responses to the immunization (data not shown). Figure 6 depicts an example CFU enumeration of bacteria recovered from respiratory tract tissues of an immunized mouse, using serial dilutions of B. pertussis tissue homogenates plated on 10% BG + Sm plates. Raw counts were multiplied by the dilution factor and total volume of the tissue lysate and the data were log transformed. CFUs from immunized animals are then compared to non-immunized (naïve) animals to determine the protective effect of the vaccine.
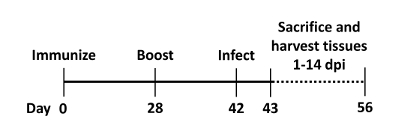
Figure 1: Representative vaccine schedule. Mice are immunized on day 0 and then boosted ~4 weeks later (d28) with the appropriate aPV. Infection of the mice with B. pertussis occurs ~2 weeks (d42) after boosting mice. After infection the animals are killed on various days (d43-56) post-inoculation. Tissue and blood is collected for downstream analysis (e.g., CFU enumeration, cytokine and antibody ELISAs). Please click here to view a larger version of this figure.

Figure 2: Anesthesia machine setup. Shown is an anesthetic vaporizer with attached induction chamber and scavenger system (inside biological safety cabinet). Please click here to view a larger version of this figure.
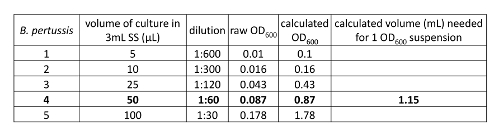
Figure 3: Preparation of 1 OD600 bacterial suspension. OD600 values were obtained from diluted primary B. pertussis cultures. One culture in log phase was used to calculate volume needed to obtain a culture at 1 OD600 culture for infection. Briefly, an OD600 of 1 was divided by the calculated OD600 of the desired culture to obtain a volume equaling 1 OD to be used for infection. Briefly, an OD600 of 1 was divided by the calculated OD600 of the desired culture to obtain a volume equaling 1 OD to be used for infection. Please click here to view a larger version of this figure.
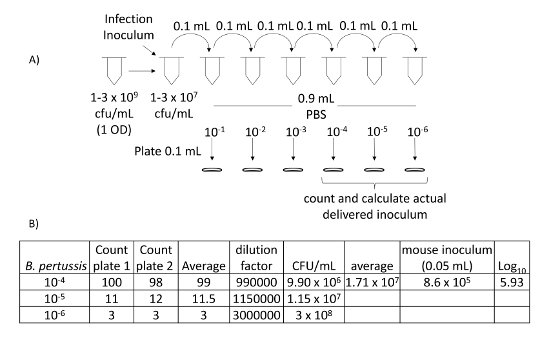
Figure 4: Calculation of delivered intranasal inoculum. (A) Schematic of serial dilution of infection inoculum that is diluted to 10-4, 10-5, and 10-6. These dilutions are plated on 10% BG + Sm, incubated for 4 days at 37 °C and then counted. (B) Counts from 10-4, 10-5, and 10-6 dilutions are then used to calculate intranasally delivered CFUs. Please click here to view a larger version of this figure.
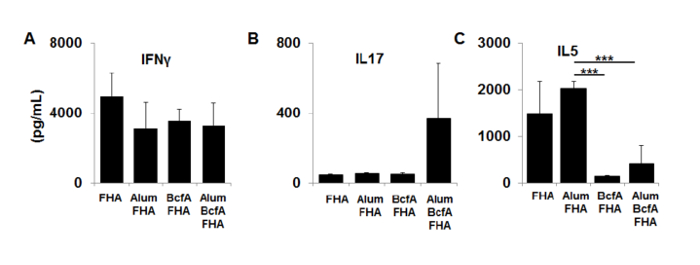
Figure 5: Cytokine production by spleen cells immunized with FHA, Alum/FHA, BcfA/FHA, and Alum/BcfA/FHA. Dissociated cells were cultured with FHA for 7 days. Supernatants were tested by sandwich ELISA for (A) IFN-γ, (B) IL-17, and (C) IL-5. Errors bars are expressed as standard deviation of the mean of each group, n = 5 per group. ***p < 0.001. The figure is adapted from a previous publication10. Please click here to view a larger version of this figure.
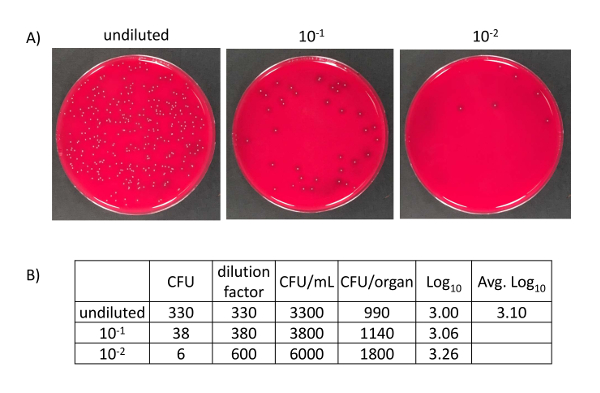
Figure 6: B. pertussis CFUs and calculations from harvested tissue from mice. (A) B. pertussis CFUs from homogenized mouse tissue. 0.1 mL serial dilutions incubated at 37 °C for 4 days. (B) Calculations based upon CFUs obtained from homogenized trachea. CFUs were multiplied by the respective dilution and then multiplied by 10 to achieve CFUs per mL. In order to obtain CFU per organ, the CFUs per mL were multiplied by the total volume in which the organ was homogenized (0.3 mL). The CFUs per organ were then log transformed. Please click here to view a larger version of this figure.
Discussion
The comprehensive protocol described here to study vaccine-induced immunity to B. pertussis infection will also permit evaluation of host responses to a variety of other pathogens. The protocol discusses methods to deliver immunizations, determine vaccine efficacy following pathogen challenge, and parallel dissection of immune function. In adapting the protocol in order to study other pathogens, several parameters would need to be modified. These include, but are not limited to, the mode of animal anesthesia, vaccine composition, dose, and route of administration. In addition, the dose and route of administration of the challenged pathogen of interest, selected tissue for harvest, and downstream evaluation of immune responses are factors that may be modified for the pathogen/disease of interest.
The mouse model of B. pertussis infection is widely used and is an excellent model for pathogenesis and vaccinology studies16,17,18,19,20. Murine models exhibit many similarities to human infection, including: i) bacteria are limited to the respiratory tract and multiply rapidly, ii) young animals display comparatively severe infections, iii) good correspondence between vaccines that protect children against pertussis and those that protect mice against infection, and iv) B. pertussis-specific T-cells and antibodies mediate natural and vaccine-induced protective immunity21,22,23,24. Therefore, the murine model permits the study of immunization effects on bacterial clearance25. Another advantage of using mice to model B. pertussis infections is the availability of genetically modified knockout and transgenic animals to study critical players in vaccine-induced immune responses19,26.
The majority of immunizations are administered parenterally, specifically via the intramuscular (i.m.) route. This route is preferred because it elicits systemic immunity with minimal local reactogenicity27,28,29. Alternatives to intramuscular injections include subcutaneous (s.c.) or intraperitoneal (i.p.) delivery, which also induce systemic responses. Subcutaneous routes are also attractive, as there is a large population of resident antigen presenting cells (APC) within the dermis with access to the vascular and lymphatic systems30. However, intradermal delivery has not been explored for the pertussis vaccine. Intraperitoneal delivery of experimental aPV generates similar immunity to intramuscular immunization31 and is a reliable injection route for murine studies, albeit impractical for clinical use.
This protocol utilizes the i.m. immunization route, which provides protection in the lungs, but not the nasopharynx32. Recent studies have shown that an intranasal (i.n.) vaccine promotes a local, mucosal immune response that protects the upper and lower respiratory tracts, particularly the nose, which serves as a reservoir of bacteria that may be subsequently transmitted to other hosts33. In addition, i.n. immunization has been shown to generate tissue resident memory that is not generated by systemic immunization33,34. Therefore, the choice of vaccine administration route depends greatly on the type of vaccine and immune response required to protect against disease.
The intranasal route of bacterial delivery described in this protocol is a widely used method for respiratory pathogens that safely and consistently delivers a bacterial inoculum directly into the respiratory tract of anesthetized mice. Our protocol uses a high-volume dose (40-50 µL) that is inhaled into the nasopharynx and travels into the lower respiratory tract. A low volume inoculum (10-20 µL) would colonize the nasopharynx, but colonization of the lungs will be minimal35. However, inhalation of B. pertussis-containing air droplets is considered the natural mode of infection of individuals and transmission. This route of administration has been used in various animal models36,37. To achieve comparable levels of infection to direct i.n. inoculation, considerably higher concentrations of the bacterial inoculum (109-1011 CFUs/mL) and delivery time are required38.
To establish colonization of mouse respiratory tract, this protocol uses fresh bacteria, grown in liquid culture to ensure that bacteria used for inoculation are in the virulent phase and are replicating in the log stage. Experimenters can also use an inoculum from pre-titrated, frozen stocks. This enables an knowledge of the CFUs being administered to the mouse. However, the bacteria in these inoculums may be in the non-virulent phase, which can reduce the efficiency of respiratory tract colonization39. After the infection, the inoculum is plated in order to determine the bacterial CFUs delivered to the mouse. Investigators may also choose to plate the inoculum prior to the in vivo infection to ensure viability of the bacteria and confirm dose of the inoculum.
After infection, bacterial CFUs are enumerated by homogenizing isolated tissue from sacrificed mice at a predetermined timepoint. A disadvantage to this method is its inability to kinetically track pathogen CFUs for a specified set of mice. Researchers must use multiple groups of animals sacrificed at different timepoints to examine vaccine-induced bacterial restriction. The number of mice in each experiment will vary depending on the number of timepoints to examine and the desired effect size. This may introduce increased biological variation. Integration of a bioluminescent or fluorescent reporter gene into the pathogen of interest will permit non-invasive monitoring of growth and dissemination in vivo throughout the course of infection35. This method reduces biological variation and minimizes the required number of experimental animals, since longitudinal data are obtained from the same group of animals.
The tissue dissociation and homogenization protocol employed here for respiratory tract tissues is also applicable for colony enumeration of other solid tissues such as liver, kidney, intestine, and/or bladder to interrogate sites of infection and dissemination of pathogens of interest.
Along with CFU enumeration, this protocol enables characterization of immune responses elicited by immunization and natural infection. Specifically, quantification of cytokines produced systemically and locally allow for the determination of efficacious immune profiles resulting from immunization. Cytokines are immunomodulators that promote cellular influx to affected tissues and activation of cellular immunity, as well as provide help for generation of humoral responses10,40,41. Using this protocol, cytokines produced in various tissue compartments, such as the lung and spleen, are detected. Although not shown here, the stimulation protocol is also applicable for evaluation of responses in the draining lymph nodes.
ELISA analysis allows detection and quantification of cytokines in media supernatants or mixed suspensions (i.e., homogenates or serum), yielding information at the population level and characterizing antigen-specific cytokine responses from a mixed cell culture at designated timepoints. In contrast, methods like ELISPOT or flow cytometry permit evaluation of the number of cells that produce an analyte and the level of expression per cell42,43. Although not described here, these methods are also applicable to detection of antigen-specific B cells. The combination of CFU enumeration and immunological assays provide complete picture of the response to immunization.
Other analyses that may be performed using this infection protocol include epigenetic or transcriptomic analysis when DNA and RNA is obtained from tissues of interest44. Thus, with consideration of the appropriate vaccine composition, immunization, and pathogen delivery route, this protocol is versatile in its implementation and easily suited for various models of infection. These techniques are also applicable to non-infectious diseases (i.e., cancer, multiple sclerosis, asthma, autoimmune diseases) where vaccines are used as a therapeutic modality.
Divulgations
The authors have nothing to disclose.
Acknowledgements
This work was supported by 1R01AI125560-01 and start-up funds from The Ohio State University.
Materials
| 2L induction chamber | Vet Equip | 941444 | |
| Fluriso | Vet One | V1 501017 | any brand is appropriate |
| Bordet Gengou Agar Base | BD bioscience | 248200 | |
| Casein | Sigma | C-7078 | |
| Casamino acids | VWR | J851-500G | Strainer Scholte (SS) media components |
| L-Glutamic acid | Research Products Int | G36020-500 | |
| L-Proline | Research Products Int | P50200-500 | |
| Sodium Chloride | Fisher | BP358-10 | |
| Potassium Phosphate monobasic | Fisher | BP362-1 | |
| Potassium Chloride | Fisher | P217-500 | |
| Magnesium Chloride hexahydrate | Fisher | M2670-500G | |
| Calcium Chloride | Fisher | C75-500 | |
| Tris base | Fisher | BP153-1 | |
| L-cysteine HCl | Fisher | BP376-100 | SS media suplements |
| Ferrous Sulfate heptahydrate | Sigma | F-7002 | |
| Niacin | Research Products Int | N20080-100 | |
| Glutathione | Research Products Int | G22010-25 | |
| Ascorbic acid | Research Products Int | A50040-500 | |
| RPMI 1640 | ThermoFisher Scientific | 11875093 | |
| FBS | Sigma | F2442-500mL | any US source, non-heat inactivated |
| gentamicin | ThermoFisher Scientific | 15710064 | |
| B-mercaptoethanol | Fisher | BP176-100 | |
| 15mL dounce tissue grinder | Wheaton | 357544 | any similar brand is appropriate |
| Cordless Hand Homogenizer | Kontes/Sigma | Z359971-1EA | any similar brand is appropriate |
| Instruments – scissors, curve scissors, forceps, fine forceps, triangle spreaders | any brand is appropriate | ||
| 3mL syringes | BD bioscience | 309657 | |
| 15mL conical tubes | Fisher | 339651 | |
| 1.5mL microfuge tubes | Denville | C2170 | |
| 70um cell strainers | Fisher | 22363548 | |
| 60mm plates | ThermoFisher Scientific | 130181 | |
| 48-well tissue culture plates | ThermoFisher Scientific | 08-772-1C | |
| 1mL insulin syringe 28G1/2 | Fisher Scientific/Excel Int. | 14-841-31 | |
| Mouse IFN-gamma ELISA Ready-SET-Go! Kit | Invitrogen / eBioscience | 50-173-21 | |
| Mouse IL-17 ELISA Ready-SET-Go! Kit | Invitrogen / eBioscience | 50-173-77 | |
| Mouse IL-5 ELISA Ready-SET-Go! Kit | Invitrogen / eBioscience | 50-172-09 |
References
- Tacken, P. J., Figdor, C. G. Targeted antigen delivery and activation of dendritic cells in vivo: steps towards cost effective vaccines. Seminars in Immunology. 23 (1), 12-20 (2011).
- Kilgore, P. E., Salim, A. M., Zervos, M. J., Schmitt, H. J. Pertussis: Microbiology, Disease, Treatment, and Prevention. Clinical Microbiology Reviews. 29 (3), 449-486 (2016).
- Dorji, D., et al. Bordetella Pertussis virulence factors in the continuing evolution of whooping cough vaccines for improved performance. Medical Microbiology and Immunology. 207 (1), 3-26 (2018).
- Feldstein, L. R., et al. Global Routine Vaccination Coverage, 2016. Morbidity and Mortality Weekly Report. 66 (45), 1252-1255 (2017).
- Cherry, J. D. Epidemic pertussis in 2012–the resurgence of a vaccine-preventable disease. New England Journal of Medicine. 367 (9), 785-787 (2012).
- Celentano, L. P., et al. Resurgence of pertussis in Europe. The Pediatric Infectious Disease Journal. 24 (9), 761-765 (2005).
- McNabb, S. J., et al. Summary of notifiable diseases. Morbidity and Mortality Weekly Report p. 54 (53), 1-92 (2007).
- Sealey, K. L., Belcher, T., Preston, A. Bordetella pertussis epidemiology and evolution in the light of pertussis resurgence. Infection, Genetics, and Evolution. 40, 136-143 (2016).
- Warfel, J. M., Merkel, T. J. The baboon model of pertussis: effective use and lessons for pertussis vaccines. Expert Reviews of Vaccines. 13 (10), 1241-1252 (2014).
- Jennings-Gee, J., et al. The adjuvant Bordetella Colonization Factor A attenuates alum-induced Th2 responses and enhances Bordetella pertussis clearance from mouse lungs. Infection and Immunity. , (2018).
- Sukumar, N., Mishra, M., Sloan, G. P., Ogi, T., Deora, R. Differential Bvg phase-dependent regulation and combinatorial role in pathogenesis of two Bordetella paralogs, BipA and BcfA. Journal of Bacteriology. 189 (10), 3695-3704 (2007).
- Stainer, D. W., Scholte, M. J. A simple chemically defined medium for the production of phase I Bordetella pertussis. Journal of General Microbiology. 63 (2), 211-220 (1970).
- Bordet, J. Le microbe de le coqueluche. Annales de l’Institut Pasteur. 20, 731-741 (1906).
- Cook, M. J. . The Anatomy of the Laboratory Mouse. , (1965).
- Sutton, S. Accuracy of Plate Counts. Journal of Validation Techniques. 17 (3), 42-46 (2011).
- Conover, M. S., Sloan, G. P., Love, C. F., Sukumar, N., Deora, R. The Bps polysaccharide of Bordetella pertussis promotes colonization and biofilm formation in the nose by functioning as an adhesin. Molecular Microbiology. 77 (6), 1439-1455 (2010).
- Cattelan, N., Jennings-Gee, J., Dubey, P., Yantorno, O. M., Deora, R. Hyperbiofilm Formation by Bordetella pertussis Strains Correlates with Enhanced Virulence Traits. Infection and Immunity. 85 (12), (2017).
- Andreasen, C., Carbonetti, N. H. Pertussis toxin inhibits early chemokine production to delay neutrophil recruitment in response to Bordetella pertussis respiratory tract infection in mice. Infection and Immunity. 76 (11), 5139-5148 (2008).
- Mills, K. H., Gerdts, V. Mouse and pig models for studies of natural and vaccine-induced immunity to Bordetella pertussis. Journal of Infectious Diseases. 209, 16-19 (2014).
- Dunne, A., et al. A novel TLR2 agonist from Bordetella pertussis is a potent adjuvant that promotes protective immunity with an acellular pertussis vaccine. Mucosal Immunology. 8 (3), 607-617 (2015).
- Denoel, P., Godfroid, F., Guiso, N., Hallander, H., Poolman, J. Comparison of acellular pertussis vaccines-induced immunity against infection due to Bordetella pertussis variant isolates in a mouse model. Vaccine. 23 (46-47), 5333-5341 (2005).
- Marr, N., et al. Protective activity of the Bordetella pertussis BrkA autotransporter in the murine lung colonization model. Vaccine. 26 (34), 4306-4311 (2008).
- Feunou, P. F., Bertout, J., Locht, C. T- and B-cell-mediated protection induced by novel, live attenuated pertussis vaccine in mice. Cross protection against parapertussis. PLoS One. 5 (4), 10178 (2010).
- Mills, K. H., Ryan, M., Ryan, E., Mahon, B. P. A murine model in which protection correlates with pertussis vaccine efficacy in children reveals complementary roles for humoral and cell-mediated immunity in protection against Bordetella pertussis. Infection and Immunity. 66 (2), 594-602 (1998).
- Higgs, R., Higgins, S. C., Ross, P. J., Mills, K. H. Immunity to the respiratory pathogen Bordetella pertussis. Mucosal Immunology. 5 (5), 485-500 (2012).
- Alving, C. R. Design and selection of vaccine adjuvants: animal models and human trials. Vaccine. 20, 56-64 (2002).
- Ipp, M. M., et al. Adverse reactions to diphtheria, tetanus, pertussis-polio vaccination at 18 months of age: effect of injection site and needle length. Pediatrics. 83 (5), 679-682 (1989).
- Fessard, C., Riche, O., Cohen, J. H. Intramuscular versus subcutaneous injection for hepatitis B vaccine. Vaccine. 6 (6), 469 (1988).
- Bergeson, P. S., Singer, S. A., Kaplan, A. M. Intramuscular injections in children. Pediatrics. 70 (6), 944-948 (1982).
- Zhang, L., Wang, W., Wang, S. Effect of vaccine administration modality on immunogenicity and efficacy. Expert Review of Vaccines. 14 (11), 1509-1523 (2015).
- Ross, P. J., et al. Relative Contribution of Th1 and Th17 Cells in Adaptive Immunity to Bordetella pertussis: Towards the Rational Design of an Improved Acellular Pertussis Vaccine. PLoS Pathogens. 9 (4), 1003264 (2013).
- Warfel, J. M., Zimmerman, L. I., Merkel, T. J. Acellular pertussis vaccines protect against disease but fail to prevent infection and transmission in a nonhuman primate model. Proceedings of the National Academy of Sciences USA. 111 (2), 787-792 (2014).
- Allen, A. C., et al. Sustained protective immunity against Bordetella pertussis nasal colonization by intranasal immunization with a vaccine-adjuvant combination that induces IL-17-secreting TRM cells. Mucosal Immunology. , (2018).
- Solans, L., et al. IL-17-dependent SIgA-mediated protection against nasal Bordetella pertussis infection by live attenuated BPZE1 vaccine. Mucosal Immunology. , (2018).
- Miller, M. A., et al. Visualization of murine intranasal dosing efficiency using luminescent Francisella tularensis: effect of instillation volume and form of anesthesia. PLoS One. 7 (2), 31359 (2012).
- Sato, Y., Izumiya, K., Sato, H., Cowell, J. L., Manclark, C. R. Aerosol infection of mice with Bordetella pertussis. Infection and Immunity. 29 (1), 261-266 (1980).
- Warfel, J. M., Beren, J., Merkel, T. J. Airborne transmission of Bordetella pertussis. Journal of Infectious Diseases. 206 (6), 902-906 (2012).
- Scanlon, K. M., Snyder, Y. G., Skerry, C., Carbonetti, N. H. Fatal Pertussis in the Neonatal Mouse Model Is Associated with Pertussis Toxin-Mediated Pathology beyond the Airways. Infection and Immunity. 85 (11), (2017).
- Martinez de Tejada, G., et al. Neither the Bvg- phase nor the vrg6 locus of Bordetella pertussis is required for respiratory infection in mice. Infection and Immunity. 66 (6), 2762-2768 (1998).
- Higgins, S. C., Jarnicki, A. G., Lavelle, E. C., Mills, K. H. TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: role of IL-17-producing T-cells. Journal of Immunology. 177 (11), 7980-7989 (2006).
- Mahon, B. P., Brady, M. T., Mills, K. H. Protection against Bordetella pertussis in mice in the absence of detectable circulating antibody: implications for long-term immunity in children. Journal of Infectious Diseases. 181 (6), 2087-2091 (2000).
- Karlsson, A. C., et al. Comparison of the ELISPOT and cytokine flow cytometry assays for the enumeration of antigen-specific T-cells. Journal of Immunological Methods. 283 (1-2), 141-153 (2003).
- Hagen, J., et al. Comparative Multi-Donor Study of IFNgamma Secretion and Expression by Human PBMCs Using ELISPOT Side-by-Side with ELISA and Flow Cytometry Assays. Cells. 4 (1), 84-95 (2015).
- Raeven, R. H. M., et al. Molecular and cellular signatures underlying superior immunity against Bordetella pertussis upon pulmonary vaccination. Mucosal Immunology. 11 (3), 1009 (2018).
