The first four steps of this protocol — the synthesis of PODS — have been designed to be robust and reliable. The deprotonation and substitution of 5-(4-aminophenyl)-1,3,4-oxadiazole-2-thiol to form the desired thioether product affords the thioether in >99% yield after just 45 minutes. Next, the ligation between 1 and N-Boc-N'-succinyl-4,7,10-trioxa-1,13-tridecanediamine was achieved via a standard peptide coupling procedure, resulting in the collection of the product (2) in 55% yield. Then, the oxidation of 2 was performed using m-chloroperoxybenzoic acid, a widely used oxidant. Following the washing steps, 3 was obtained as a pale solid in ~90% yield. Finally, the removal of the tert-butyloxycarbonyl protecting group from 3 was done according to standard procedures, using a 4:1 ratio of dichloromethane:trifluoroacetic acid. After the lyophilization of the aqueous phase, our product — PODS — was obtained as a white powder in 98% yield. The progress of the reaction was followed via thin layer chromatography, and the identity of each product was confirmed via 1H-NMR, 13C-NMR, and HRMS-ESI (Table 1).
One of the principal advantages of the PODS reagent is its modularity. A variety of chelators, fluorophores, toxins, or other cargoes can be appended to the compound's pendant amine. In the protocol at hand, we are using the ubiquitous chelator DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) as a representative payload. DOTA, of course, has been used in a wide range of biomolecular radiopharmaceuticals as a chelator for radiometals including 68Ga, 64Cu, 111In, 90Y, 177Lu, and 225Ac. To this end, an isothiocyanate-bearing variant of DOTA (p-SCN-Bn-DOTA) was employed and coupled to the pendant amine of PODS via straightforward coupling conditions. The resultant bifunctional chelator was then purified via reverse phase C18 HPLC and isolated in ~75% yield. As with the other precursors, the progress of the reaction was followed via thin layer chromatography, and the identity of the product was confirmed via 1H-NMR, 13C-NMR, and HRMS-ESI (Table 1).
In the final step of the protocol, we discuss the site-selective bioconjugation of PODS-DOTA to a model immunoglobulin, the HER2-targeting antibody trastuzumab. To this end, the disulfide linkages of the antibody's hinge region are selectively reduced with the reducing agent TCEP [tris(2-carboxyethyl)phosphine]. Following this reduction step, the antibody is incubated with PODS-DOTA for 2 h at room temperature and subsequently purified via size exclusion chromatography. In this case, the purified, DOTA-bearing immunoconjugate was obtained in ~80% yield, and MALDI-ToF analysis revealed a degree of labeling (DOL) of ~1.8 DOTA/mAb. Generally speaking, we have found that 10 equivalents of TCEP, 10 equivalents of the PODS reagent, and a 2 h incubation are sufficient to yield an immunoconjugate with a DOL of 2 PODS/mAb (Table 2). This result remains consistent across a range of human, humanized, and chimeric IgG1 antibodies; however, the same conditions produce immunoconjugates with a DOL of only ~1.5 when working with murine IgG1 antibodies. All this said, researchers should optimize these reaction conditions for new antibodies and PODS-bearing cargoes. Finally, and importantly, with respect to the final product, we have repeatedly and reproducibly found that PODS-based immunoconjugates exhibit immunoreactivities equal to or better than analogous constructs created using random or maleimide-based conjugation strategies.
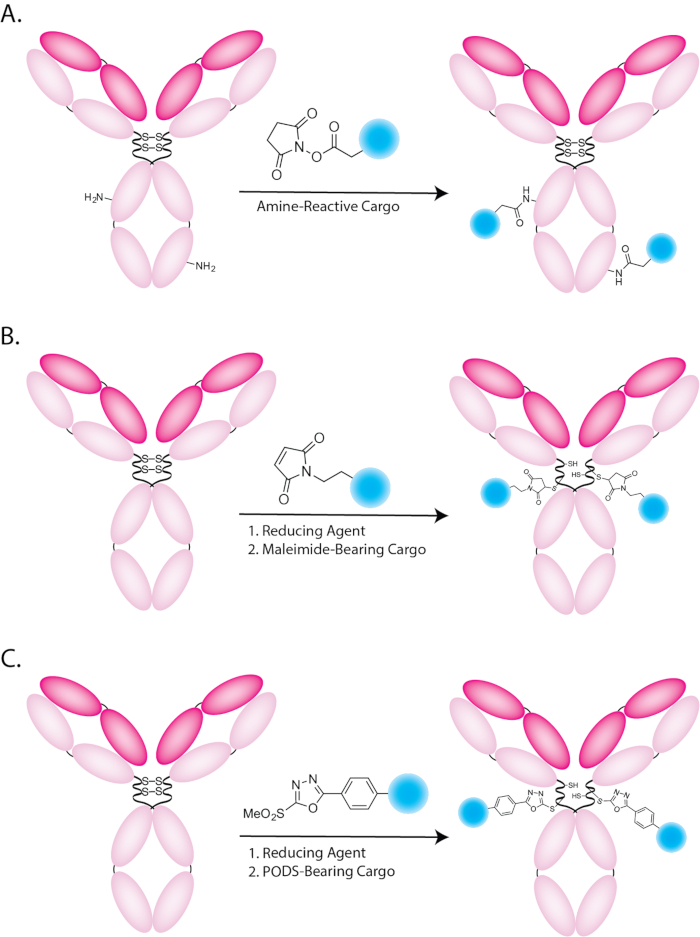
Figure 1: Schematic illustration of bioconjugations using (A) amine-reactive, (B) maleimide-bearing, and (C) PODS-bearing cargoes. Please click here to view a larger version of this figure.
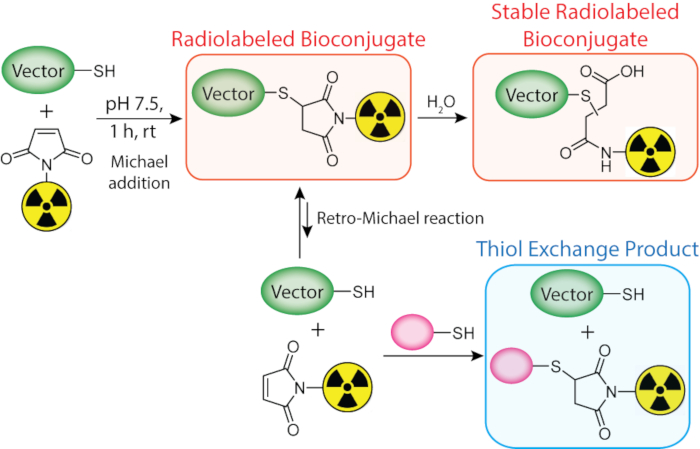
Figure 2: The Michael addition of a thiol-bearing biomolecule (green) and a radionuclide-bearing maleimide (yellow) to form a radiolabeled bioconjugate, as well as the additional reactions that the radiolabeled construct can undergo in the presence of endogenous thiol-bearing molecules (pink). RT = Room temperature. Figure reprinted with permission from Adumeau, P., Davydova, M., Zeglis, B. M. Thiol-Reactive Bifunctional Chelators for the Creation of Site-Selectively Modified Radioimmunoconjugates with Improved Stability. Bioconjugate Chemistry. 29, 1364-1372 (2018). Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
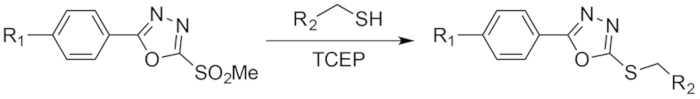
Figure 3: Schematic of the reaction between PODS and a thiol. Figure reprinted with permission from Adumeau, P., Davydova, M., Zeglis, B. M. Thiol-Reactive Bifunctional Chelators for the Creation of Site-Selectively Modified Radioimmunoconjugates with Improved Stability. Bioconjugate Chemistry. 29, 1364-1372 (2018). Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
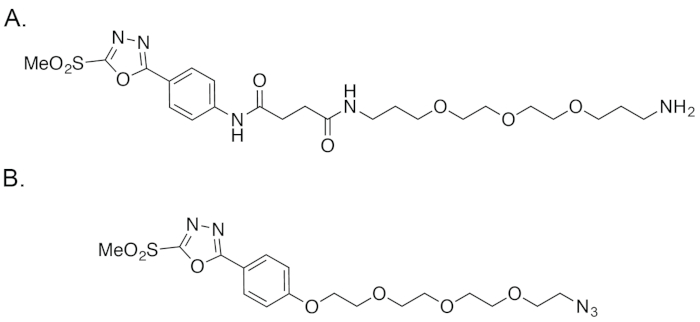
Figure 4: The structure of PODS (A) as well as (B) the reagent reported by Barbas, et al.18,19 Please click here to view a larger version of this figure.
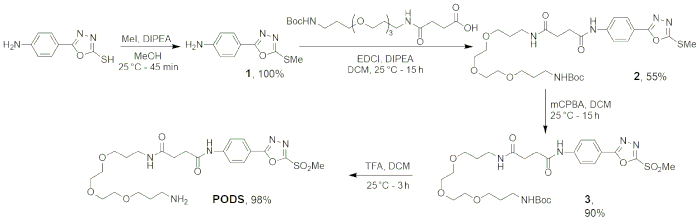
Figure 5: Scheme of the four-step synthesis of PODS. Figure reprinted with permission from Adumeau, P., Davydova, M., Zeglis, B. M. Thiol-Reactive Bifunctional Chelators for the Creation of Site-Selectively Modified Radioimmunoconjugates with Improved Stability. Bioconjugate Chemistry. 29, 1364-1372 (2018). Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
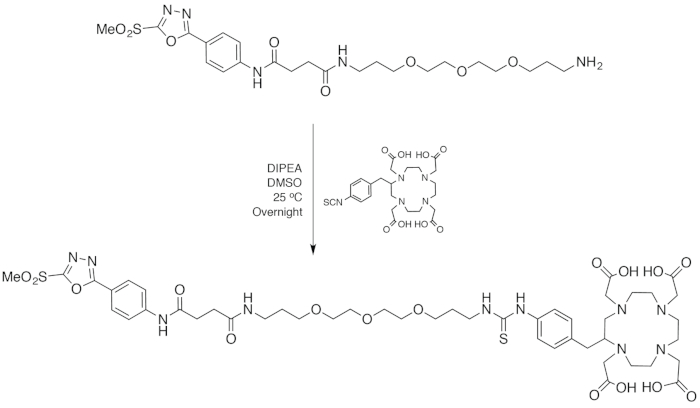
Figure 6: Scheme of the synthesis of PODS-DOTA. Please click here to view a larger version of this figure.
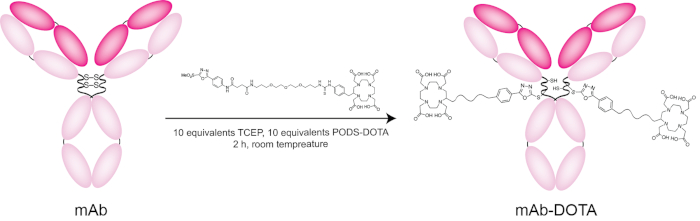
Figure 7: Scheme of the bioconjugation of trastuzumab with PODS-DOTA. Please click here to view a larger version of this figure.
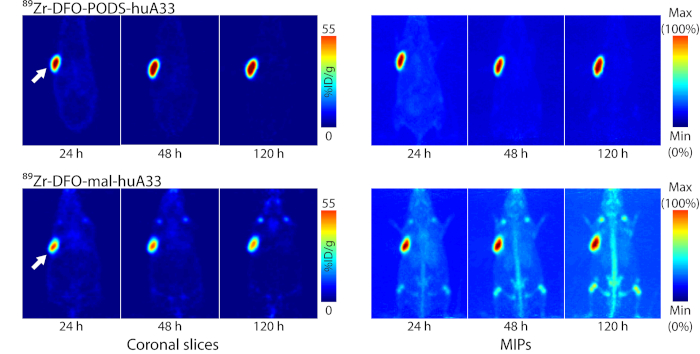
Figure 8: Comparison of the in vivo behavior of 89Zr-labeled radioimmunoconjugates of huA33 created using PODS-based (89Zr-DFO-PODS-huA33) and maleimide-based (89Zr-DFO-mal-huA33) bioconjugation strategies. Planar (left) and maximum intensity projection (right) PET images of athymic nude mice bearing A33 antigen-expressing SW1222 colorectal cancer xenografts (white arrow) following the injection of 89Zr-DFO-PODS-huA33 and 89Zr-DFO-mal-huA33 (140 µCi, 60-65 µg). The coronal slices intersect the center of the tumors. Figure reprinted with permission from Adumeau, P., Davydova, M., Zeglis, B. M. Thiol-Reactive Bifunctional Chelators for the Creation of Site-Selectively Modified Radioimmunoconjugates with Improved Stability. Bioconjugate Chemistry. 29, 1364-1372 (2018). Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
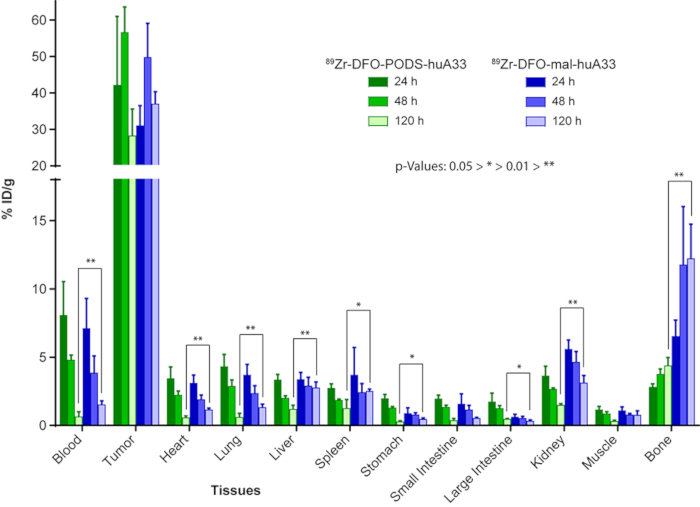
Figure 9: Comparison of the in vivo behavior of 89Zr-labeled radioimmunoconjugates of huA33 created using PODS-based (89Zr-DFO-PODS-huA33) and maleimide-based (89Zr-DFO-mal-huA33) bioconjugation strategies. Biodistribution data after the administration of 89Zr-DFO-PODS-huA33 and 89Zr-DFO-mal-huA33 (30 µCi, 15-18 µg) to athymic nude mice bearing A33 antigen-expressing subcutaneous SW1222 human colorectal cancer xenografts. The values for the stomach, small intestine, and large intestine include contents. Figure reprinted with permission from Adumeau, P., Davydova, M., Zeglis, B. M. Thiol-Reactive Bifunctional Chelators for the Creation of Site-Selectively Modified Radioimmunoconjugates with Improved Stability. Bioconjugate Chemistry. 29, 1364-1372 (2018). Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
| Compound | 1H-NMR Shifts | 13C-NMR Shifts | HRMS | |||
| 1 | (500 MHz, CDCl3) 7.79 (2H, d, J = 8.5 Hz), 6.72 (2H, d, J = 8.5 Hz), 4.04 (2H, br s), 2.75 (3H, s) | (125 MHz, CDCl3) 166.3, 163.7, 149.7, 128.5, 114.8, 113.5, 14.8 | m/z Calcd for [C9H9N3OS+H]+: 208.0539; found: 208.0539; Δ: 0.0 ppm | |||
| 2 | (500 MHz, CDCl3) 9.68 (1H, s), 7.91 (2H, d, J = 9.0 Hz), 7.71 (2H, d, J = 8.5 Hz), 6.82 (1H, s), 4.99 (1H, s), 3.70-3.45 (12H, m), 3.41 (2H, q, J = 6.0 Hz), 3.20 (2H, q, J = 6.5 Hz), 2.76 (3H, s), 2.71 (2H, m), 2.63 (2H, m), 1.80-1.70 (4H, m), 1.42 (9H, s) | (125 MHz, CDCl3) 172.6, 171.3, 165.8, 164.6, 156.2, 141.8, 127.7, 119.6, 118.6, 79.2, 70.6, 70.5, 70.3, 70.1, 69.6, 38.8, 38.5, 33.5, 31.6, 29.9, 28.6, 14.8 | m/z Calcd for [C28H43N5O8S+Na]+: 632.2725; found: 632.2722; Δ: -0.47 ppm | |||
| 3 | (500 MHz, CDCl3) 9.99 (1H, s), 7.98 (2H, d, J = 9.0 Hz), 7.75 (2H, d, J = 8.5 Hz), 6.88 (1H, s), 4.99 (1H, s), 3.66-3.50 (15H, m), 3.41 (2H, q, J = 6.0 Hz), 3.20 (2H, q, J = 6.5 Hz), 2.71 (2H, m), 2.65 (2H, m), 1.80-1.70 (4H, m), 1.43 (9H, s) | (125 MHz, CDCl3) 172.6, 171.5, 166.5, 161.6, 156.1, 143.4, 128.7, 119.6, 116.4, 79.1, 70.5, 70.4, 70.2, 70.0, 69.4, 43.0, 38.8, 38.4, 33.2, 31.3, 29.7, 28.4 | m/z Calcd for [C28H43N5O10S+H]+: 642.2803; found: 642.2797; Δ: -0.93 ppm | |||
| PODS | (500 MHz, D2O) 7.85 (2H, d, J = 9.0 Hz), 7.55 (2H, d, J = 8.5 Hz), 3.60-3.45 (15H, m), 3.45 (2H, t, J = 6.5 Hz), 3.20 (2H, t, J = 6.5 Hz), 3.04 (2H, t, J = 7.0 Hz), 2.67 (2H, t, J = 6.5 Hz), 2.54 (2H, t, J = 6.5 Hz), 1.87 (2H, qt, J = 6.5 Hz), 1.70 (2H, qt, J = 6.5 Hz) | (125 MHz, D2O) 174.5, 173.2, 166.8, 161.4, 142.2, 128.6, 120.3, 116.6, 69.4, 69.4, 69.3, 69.2, 68.2, 68.2, 42.5, 37.6, 36.2, 31.9, 30.7, 28.2, 26.4 | m/z Calcd for [C23H35N5O8S+H]+: 542.2279; found: 542.2281; Δ: 0.37 ppm | |||
| PODS-DOTA | (600 MHz, DMSO-d6) 10.46 (1H, s), 9.74 (1H, bs), 8.04 (2H, d, J = 8.6 Hz), 7.99 (1H, s), 7.90 (1H, t, J = 5.0 Hz), 7.86 (2H, d, J = 6.5 Hz), 7.44 (2H, d, J = 7.9 Hz), 7.24 (2H, d, J = 7.1 Hz), 4.35-2.41 (45H, m), 3.70 (3H, s), 1.76 (2H, q, J = 6.3 Hz), 1.61 (2H, q, J = 6.5 Hz) | (125 MHz, DMSO-d6) 171.8, 171.4, 166.1, 162.2, 158.8, 158.6, 129.8, 129.0, 127.6, 123.3, 119.5, 118.5, 116.5, 116.4, 70.2, 70.1, 70.0, 68.7, 68.5, 43.4, 41.8, 36.3, 32.2, 30.4, 29.8, 29.1 | m/z Calcd for [C47H68N10O16S2+H]+: 1093.4334; found: 1093.4327; Δ: -0.64 ppm | |||
Table 1. Characterization data for the synthetic intermediates described as well as PODS and PODS-DOTA.
| Antibody | Type | Constant Region | Ratio of PODS:mAb |
| Human plasma IgG | Human | Human IgG | 2.1 ± 0.1 |
| Trastuzumab | Humanized | Human IgG1 | 2.0 ± 0.1 |
| huA33 | Humanized | Human IgG1 | 2.1 ± 0.1 |
| Cetuximab | Chimeric | Human IgG1 | 2.2 ± 0.1 |
| AR 9.6 | Murine | Murine IgG1 | 1.4 ± 0.1 |
| Mouse plasma IgG | Murine | Murine IgG | 1.5 ± 0.1 |
Table 2. Degree of labeling of different antibodies following conjugation with a PODS-bearing fluorophore. Values are shown standard deviations. Table reprinted with permission from Adumeau, P., Davydova, M., Zeglis, B. M. Thiol-Reactive Bifunctional Chelators for the Creation of Site-Selectively Modified Radioimmunoconjugates with Improved Stability. Bioconjugate Chemistry. 29, 1364-1372 (2018). Copyright 2018 American Chemical Society.
