A High-throughput Assay to Assess and Quantify Neutrophil Extracellular Trap Formation
Summary
This protocol describes a highly sensitive and high throughput neutrophil extracellular trap (NET) assay for the semi-automated quantification of ex vivo NET formation by immunofluorescence three-dimensional confocal microscopy. This protocol can be used to evaluate NET formation and degradation after different stimuli and can be used to study potential NET-targeted therapies.
Abstract
Neutrophil extracellular traps (NETs) are immunogenic extracellular DNA structures that can be released by neutrophils upon a wide variety of triggers. NETs have been demonstrated to serve as an important host defense mechanism that traps and kills microorganisms. On the other hand, they have been implicated in diverse systemic autoimmune diseases. NETs are immunogenic and toxic structures that contain a pool of relevant autoantigens including anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis (AAV) and systemic lupus erythematosus (SLE). Different forms of NETs can be induced depending on the stimulus. The amount of NETs can be quantified using different techniques including measuring DNA release in supernatants, measuring DNA-complexed with NET-molecules like myeloperoxidase (MPO) or neutrophil elastase (NE), measuring the presence of citrullinated histones by fluorescence microscopy, or flow cytometric detection of NET-components which all have different features regarding their specificity, sensitivity, objectivity, and quantity. Here is a protocol to quantify ex vivo NET formation in a highly-sensitive, high-throughput manner by using three-dimensional immunofluorescence confocal microscopy. This protocol can be applied to address various research questions about NET formation and degradation in health and disease.
Introduction
The formation of neutrophil extracellular traps (NETs) is the process in which neutrophils release their DNA in an extracellular three-dimensional (3D) web like structure, complexed with a wide range of antimicrobial and dangerous molecules, granular and cytoplasmic enzymes, peptides and proteins. These immunogenic and toxic structures have an important physiological role in the innate immune defense of healthy individuals by trapping and killing infectious pathogens1. However, they have also been demonstrated to be involved in thrombosis2 and various systemic autoimmune diseases, including anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis (AAV)3, systemic lupus erythematosus (SLE)4,5, antiphospholipid syndrome (APS)2,6, rheumatoid arthritis (RA), psoriasis, and gout7,8,9.
In vitro NET formation has been widely studied with the chemical compound phorbol 12-myristate 13-acetate (PMA), which induces massive NET formation. However most physiological stimuli induce much lower levels of NET formation10. To study NET-triggers in, for example, an autoimmune disease setting, a standardized, sensitive, high-throughput quantitative assay is needed to detect and quantify NET formation. Quantification of NETs has proven to be challenging and is currently performed by different methods, each with their own advantages and limitations11. A commonly used method is the detection of DNA in the supernatants12, which is objective but does not discriminate between the origin of the DNA (apoptotic, necrotic, NETs), and therefore is not very specific for NETs. Secondly, enzyme-linked immunosorbent assays (ELISAs) of DNA-complexed with NET-specific proteins, for example, myeloperoxidase (MPO) or neutrophil elastase (NE), are a more specific approach to detect NETs and were demonstrated to correlate well with citrullinated histone-3 (CitH3) positive NETs13. However, it is not known whether this method is sensitive enough to pick up all NETs (e.g., MPO, NE, and CitH3 negative NETs). A third approach is immunofluorescence microscopy that is used to detect co-localization of NET-associated molecules (NE, MPO, CitH3) with extracellular DNA to quantify NETs. This method is generally specific for NETs, but it cannot be applied as a high-throughput method and is not objective due to observer bias. Additionally, this method does not take into account MPO-, NE-, CitH3-negative NETs that are frequently present depending on the used NET-trigger14,15. Flow cytometry approaches detect NETs through a changed forward/sideward scatter (FSC/SSC) indicating swelling of the nucleus in NET-ting neutrophils16. This method does not take into account the different forms of NET formation that have been identified, which might not involve swelling of the nucleus, such as vital NET formation17. Lastly, immunofluorescence confocal microscopy has been applied to visualize and quantify NET formation by directly staining extracellular DNA with a cell-impermeable dye that stains extracellular DNA12,18. Generally, 5 to 10 high-power fields are manually picked and assessed, which covers 1-5% of each well of a 96-well plate11,17. Manual selection of images is not always objective, prone to bias and not attractive for high-throughput analysis. An automated, high-throughput NET quantification assay was recently developed, which imaged 11% of the well in a 3D manner covering 13 µm through Z-stacked immunofluorescence confocal microscopy, thereby leading to a highly sensitive technique to assess NETs compared to the conventional methods10. The current report describes the most recent protocol to quantify NET formation through an automated, highly sensitive assay using 3D confocal microscopy, which achieves a total imaged area of 45% of each well and covers 27 µm through Z-stacks. This protocol is suitable to quantify, with a high sensitivity, low levels of NET formation in an objective and unbiased manner.
Protocol
All patients and healthy controls consented to participate in the LUMC biobank. Both biobanking studies were approved by the LUMC ethical committee.
1. Isolation of Healthy Neutrophils
- Obtain 20 mL of peripheral blood from a healthy donor in two 10 mL EDTA-coated tubes.
- Put 10 mL of blood in a sterile 50 mL tube and add phosphate buffered saline (PBS) up to 32.5 mL.
- Add density gradient (e.g., Ficoll-amidotrizoaat) under the cells.
- Take up 14 mL of density gradient with a 10 mL pipet and pipet controller.
- Place the pipet on the bottom of the 50 mL tube.
- Take the pipet controller off the pipet, allowing the density gradient to flow out of the pipet by gravity until the maximum is reached by capillary effect (1-2 mL will be left), without using the motor.
- Remove pipet by placing a thumb on top of the pipet, thereby preventing density gradient from leaking out during removal of the pipet.
- Spin the tubes for 20 min at 912 x g and room temperature (RT) without acceleration or brake.
NOTE: Red blood cells (RBC) and neutrophils have a high density and are at the bottom of the 50 mL tube. Peripheral blood mononuclear cells (PBMCs) are separated and on top of the density gradient as a white ring. PBS-diluted plasma will be above the PBMCs. If needed, PBMCs can be isolated by transferring the white ring to a new 50 mL tube with additional washing steps with PBS. - Carefully remove the white ring containing PBMCs first, followed by removal of the PBS-diluted plasma and lastly the density gradient layer as much as possible.
- To isolate neutrophils from the neutrophil/erythrocytes mix, lyse erythrocytes by cold sterile distilled water.
- Take cold sterile distilled water bottle and a 10x concentrated PBS flask from the fridge.
- Work quickly for this step. Add 36 mL of cold sterile distilled water directly on top of the pellet and mix once carefully. Add 4 mL of 10x PBS after 20 s to make an isotonic solution. Mix once carefully.
- Spin tube for 5 min at 739 x g and 4 °C (for the removal of RBCs). Neutrophils will be in the white pellet.
- Carefully discard the supernatant. Perform step 1.6.2 again and make sure the pellet is suspended properly.
- Spin tube for 5 min at 328 x g and 4 °C.
- Carefully remove the supernatant and resuspend the pellet in 5 mL of PBS. Count the neutrophils and keep them on ice.
NOTE: Expected yield from 1 tube of blood (10 mL) is approximately 15-75 million neutrophils.
2. Red Fluorescent Cell Labelling of Neutrophils
- Make a neutrophil suspension of 10-20 million neutrophils in 2 mL of PBS in a 15 mL tube.
- Make a solution of 2 mL PBS with 4 µL of 2 µM red fluorescent cell linker (see Table of Materials) in a different 15 mL tube. Add this gently to the neutrophil suspension and mix carefully.
- Incubate in the dark for exactly 25 min at 37 °C to label the neutrophils with the red fluorescent cell linker.
- Inactivate the labeling by adding RPMI 1640 medium containing 10% heat inactivated fetal bovine serum (FCS) at RT up to 15 mL and mix once carefully. Make sure that the pellet is carefully resuspended if a pellet has formed.
- Spin tube for 5 min at 328 x g and RT.
- Remove the supernatant and resuspend pellet in 5 mL of phenol red-free RPMI 1640 medium containing 2% FCS and 10% P/S at RT. Count the neutrophils.
NOTE: A cell loss of 50% can occur after red fluorescent cell labelling.
3. Induction of Neutrophil Extracellular Trap Formation
- Make a cell suspension of 0.42 x 106 cells/mL in phenol red free RPMI 1640 medium containing 2% FCS and 10% P/S.
- Add 37,500 neutrophils in 90 µL per well in a black 96-well, flat bottom plate.
- Add 10 µL of the chosen stimulus (e.g., sera of patient) in triplicate to reach a concentration of 10% in the well. Always include a negative control (medium) in triplicate.
- Incubate in the dark at 37 °C for the desired time, ranging from 30 min to 2, 4 or 6 h. Incubating for 4 h is suggested.
- Calculate the volume needed to add 25 µL of 5 µM impermeable DNA dye (see Table of Materials), to reach a final concentration of 1 µM in the well. Make a predilution if necessary in RPMI 1640 medium containing 2% FCS and 10% P/S to obtain a 5 µM concentration.
- Add 25 µL of 5 µM impermeable DNA dye 15 min before the end of the incubation time. Continue incubation for another 15 min at 37 °C in the dark.
- Remove the supernatant (~125 µL) very carefully and store if needed. Add 100 µL of 4% paraformaldehyde (PFA). Keep in the dark, and immediately continue with step 4.
4. NET Visualization with 3D High-content, High-resolution Immunofluorescence Confocal Microscopy
- Configure the settings on the immunofluorescence confocal microscope by clicking on the Acquisition setup.
- Click on the Configure tab.
- Select the objective and the camera. Choose the 10X Apo Lambda objective with an acquisition mode of a confocal 60 µm pinhole.
- Click on Plate and choose the 96-well plastic plate. Select the sites to visit and choose a fixed number of sites. Fill in 3 columns and 3 rows without overlap (0 µm), which cover a total of 45% of the well.
- Click on Acquisition. Select Enable Laser-Based Focusing. Select Acquire Z Series/Time Series if needed.
- Click on Site Autofocus.
- Click on Focus on Plate Bottom | Off-Set by Bottom Thickness. For the initial well to find the sample, choose the first well acquired. For the site autofocus, choose all sites.
- Click on Wavelengths.
- For the number of wavelengths, choose 2. For the TL shading correction refinement level, choose 2.
- For Wavelength 1, select Texas Red.
- For the autofocus options, select laser with Z offset, post laser offset 1.1 µm. Use Z-stack with a custom range of 200-10.
- For Wavelength 2, select FITC.
- For the autofocus options, select laser with Z offset from w1 0 µm. Use Z-stack with a custom range of 200-10.
- For the acquisition options, select Z series and 2D projection image maximum. For the acquisition options, select Shading Correction | Off.
- Select Z Series. Select the number of steps: 10. Select the step size: 3 μm (total range will be 27 µm).
- Put the plate in the immunofluorescence confocal microscopy.
- Click on the Run tab.
- Fill out plate name and description and choose the storage location.
- Select the wells that need to be acquired.
- Click on the Run tab.
- Choose exposure time for Texas Red and FITC.
- Click on Acquire Plate to start the acquisition, which will take approximately 1 h per plate.
5. Analysis of NET Formation
- Use an image processing program designed for analysis of scientific multidimensional images (see Table of Materials) to analyze NET formation.
- Transfer the acquired image data to a separate hard drive.
- Select the color adding tool.
- Select w1 and choose the folder in the hard drive where the data is stored.
- Select w2 and choose the folder in the hard drive where the data is stored.
NOTE: Use a standard macro that uses w1 in the file name to add red color to the Texas Red images and uses w2 to add green color to the FITC images.
- Select Analysis Macro.
- Select w1, choose the threshold value (intensity threshold), which is usually 10. Select the desired pixel value (size threshold, e.g., 100).
- Select w2, choose the threshold value (intensity threshold), which is usually 10. Select the desired pixel value (size threshold, e.g., 500).
- Choose the destination for the spreadsheet file, run analysis, and save log files afterwards.
- Analyze data in a spreadsheet.
Representative Results
Neutrophil extracellular trap (NET) formation is quantified in a 3D manner by quantifying stained extracellular DNA over 10 Z-stacks with 3 µm distance starting off at the focal plane in each well. By measuring the cumulative area, the sensitivity of the assay increases (Figure 1A). The isolated neutrophils have a mean purity of 98.7% with standard deviation (SD) of 1.10% measured in 14 different samples from different isolations. The mean percentage of red blood cells is 1.04% ± 1.1% SD and the mean percentage of monocytes is 0.085% ± 0.17% SD (data not shown). The total area of stained neutrophils imaged are quantified only in the focal plane in each well, which correlate significantly with the total neutrophil count in each well with a Pearson correlation coefficient of 0.99 (95% confidence interval [CI] 0.985-0.997, p < 0.0001) (Figure 1B). The representative outcome of quantification of NET formation in neutrophils stimulated with 10% sera of AAV patients or medium (MED) as a negative control, expressed as cumulative stained extracellular DNA area over 10 Z-stacks per imaged neutrophil (Figure 1C). Snapshots of representative images of unstimulated neutrophils (Figure 2A) and of NETs in AAV-stimulated neutrophils (Figure 2B) are shown.
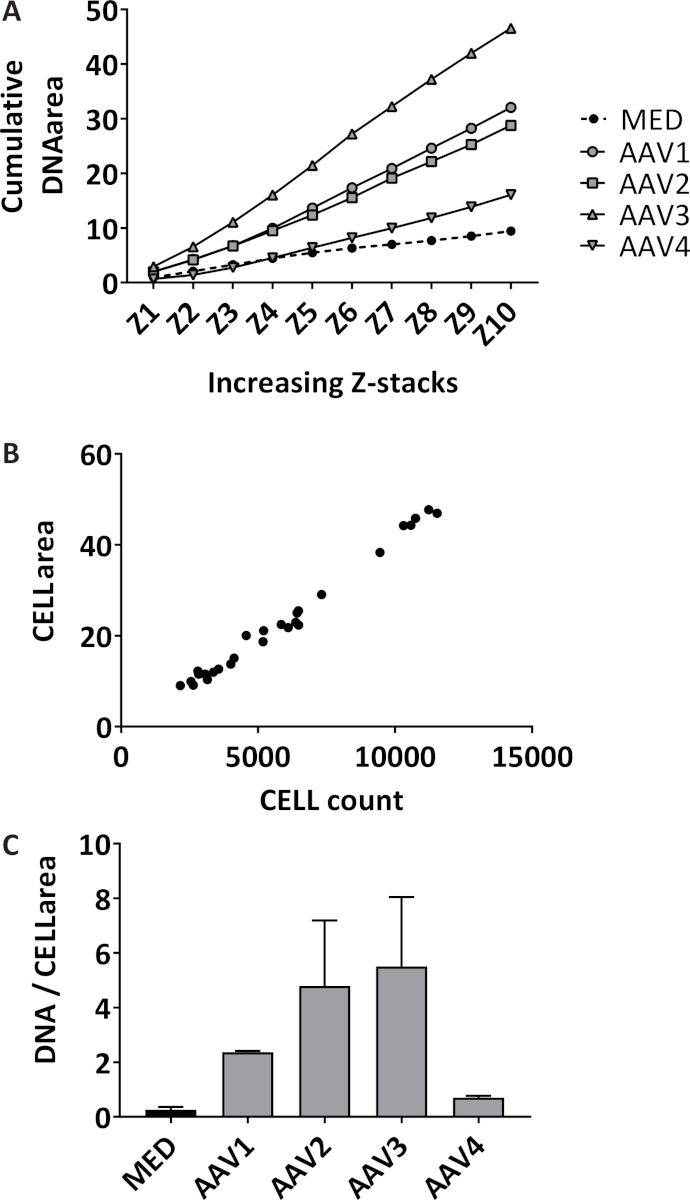
Figure 1: Quantification of NET formation by measuring extracellular DNA and neutrophil count. (A) Area is cumulatively quantified over the 10 Z-stacks for each well for unstimulated neutrophils (MED) and for neutrophils stimulated with 10% serum of ANCA-associated vasculitis (AAV) patients (n = 4) quantified with an image processing program. Each stimulus was tested in triplicate, each point represents the median value. (B) Red fluorescent labelled cell area and cell count were quantified in the focal plane of each well by the image processing program (R2 = 0.99, p < 0.0001). (C) NET formation is expressed as cumulative area per imaged neutrophil (cell area). The mean ± standard error of the mean (SEM) of each triplicate is plotted per stimulus. Please click here to view a larger version of this figure.
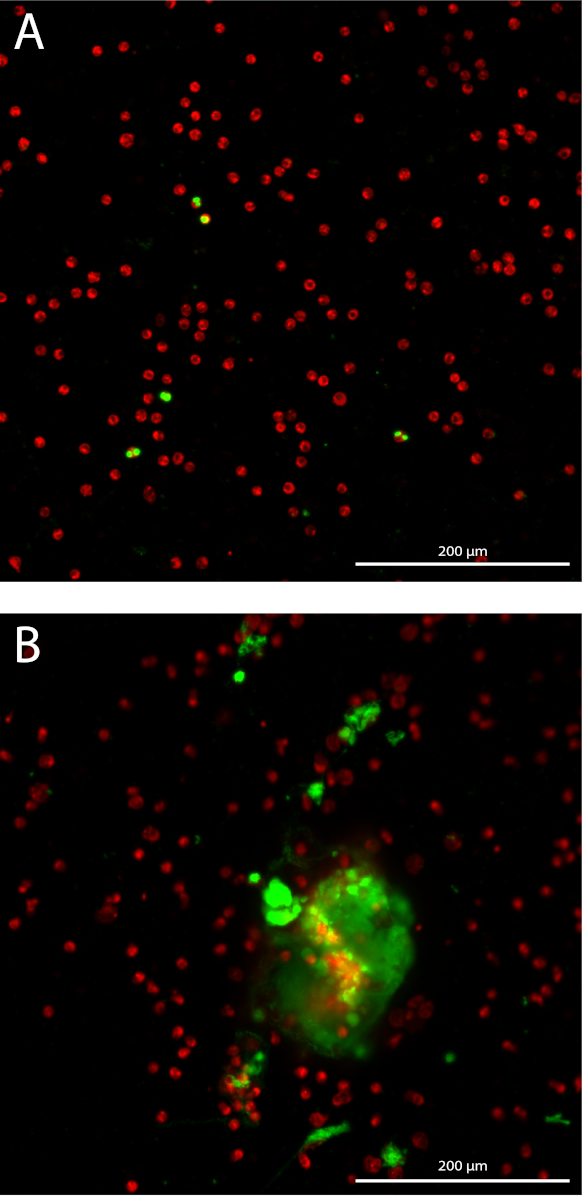
Figure 2: Snapshots of NET quantification assay. Fluorescent labelled neutrophils are shown in red, and stained extracellular DNA is shown in green. 10x Plan Apo Lambda objective. (A) Unstimulated neutrophils. (B) Neutrophils stimulated with AAV serum. Please click here to view a larger version of this figure.
Discussion
The most critical part of this assay is the need to use freshly isolated neutrophils for each experiment because neutrophils are short-lived and die when frozen. This requires a healthy donor each time, which could implicate some variations due to donor characteristics. One of these variations is the activation status of the neutrophils. Neutrophils could be activated already in vivo prior to isolation. In addition, neutrophils can be activated throughout the isolation steps notably during lysis of erythrocytes, therefore an experienced handler of neutrophils is required to minimize the activation of neutrophils. In general, isolation of neutrophils should be performed as soon as possible after blood drawing and the experiment should not be paused to avoid excessive spontaneous activation. Secondly, rough handling of neutrophils should be avoided. As such, the notable advantage of the described protocol is the minimal pipetting interventions once neutrophils are seeded in a 96-well plate. Importantly, the activation status of the neutrophils is best assessed in the unstimulated condition, in which this assay can sensitively detect low levels of NET formation. Another factor that could influence the assay is the use of FCS in the medium. The percentage of FCS has been decreased from 10% to 2% to avoid the possible suppression of NET formation by antioxidant activity19,20 or the possible activation of the neutrophils despite heat inactivation. Culture without FCS or with the use different types of media has not been tried. An unstimulated or medium control is always taken along when performing the assay to have an indication of the background signal (e.g., activation status of the neutrophils). The fold increase for each stimulus compared to the unstimulated sample is displayed to achieve consistent results over different experiments using the same stimulus.
An important factor for a possible high background is staining of extracellular DNA that is unrelated to the process of NET formation. The present assay attempts to reduce this by removing the extracellular DNA staining immediately after the short incubation period of 15 min and by analyzing the plate directly after fixation. Therefore, it is essential to use an advanced confocal microscope that has enough speed and analytical power to capture the 96-well plate within 1 to 2 h. Automated setting of the exposure time and focus is recommended. As such, the microscope setting can vary between each sample and experiment with respect to the color intensity threshold which is necessary for overall optimal picture quality. The latter influences the eventual capability to correctly quantify neutrophils and NETs and the optimal setting should therefore be confirmed by using multiple control samples (e.g., serum of healthy controls). During the analysis of captured images, the use of a pixel threshold and size threshold in the analysis program allows for a better selection of NET formation.
Extracellular DNA derived from NET formation can be the result of distinct death pathways, including NETosis, necroptosis, pyroptosis, ferroptosis or even non-lytic process of vital NET formation1. As such, a limitation of the present assay is that by staining only for extracellular DNA there is no differentiation possible between NET formation and other relevant cell death pathways. It is possible to achieve this by using either selective inhibitors of distinct death pathways to discriminate between the different forms underpinning NET formation or confirm by separate immunostainings the presence of specific NET markers, such as citH3 and NE, co-localized with DNA. The co-localization of extracellular DNA with citH3 and NE has recently been confirmed for this assay10. The advantage of avoiding NET specific markers in this assay, allows to assess all forms of NET formation leading to the extrusion of DNA by neutrophils as complete and as objective as possible with the potential of high-throughput screening. The applicability of this protocol has been shown in studying low level NET induction mediated by immune complexes in auto-immune disease in which the ability to detect qualitative and quantitative differences might be more important than the type of process involved10,21,22. Illustrating that this novel NET quantification assay can be of added value for different researchers to investigate various aspects of NET formation. Small adjustments to the assay are easily implemented: adjustment of the stimulation period, the use of a favorite NET marker to focus on one specific death pathway leading to NET formation, the use of a different magnification or the use of different NET criteria in the quantification and analysis.
In conclusion, the protocol provided is a highly-sensitive broadly applicable assay for the semi-automated quantification of NET formation for the evaluation of ex vivo induction of NETs upon different stimuli.
Divulgations
The authors have nothing to disclose.
Acknowledgements
The work of Eline J. Arends and Y.K. Onno Teng is supported by the Dutch Kidney Foundation (17OKG04), Clinical Fellowship from the Netherlands Organization for Scientific Research (90713460). Laura S. van Dam's work is supported by the Foundation for Research in Rheumatology (FOREUM).
Materials
| Aqua Sterile H2O | B. Braun, Melsungen, Germany | 12604052 | |
| Fetal bovine serum (FCS) | Bodinco, Alkmaar, The Netherlands | Used in high concentrations it could influcence NET formation | |
| Ficoll 5,7% – amidotrizoaat 9% density 1,077 g / mL | LUMC, Leiden, The Netherlands | 97902861 | |
| Immunofluorescence confocal microscope | Image Xpress Micro Confocal (Molecular Devices, Sunnyvale, CA, USA) | ||
| Neutralization PBS (10x) | Gibco, Paisley, UK | 70011-036 | |
| Penicillin / streptomycin (p/s) | Gibco, Paisley, UK | 15070063 | |
| Phenol red free RPMI 1640 (1x) | Gibco, Paisley, UK | 11835-063 | Phenol red can interfere with the immunofluorescence signal |
| Phosphate-buffered saline (PBS) | B. Braun, Melsungen, Germany | 174628062 | |
| PKH26 2 uM Red fluorescent cell linker | Sigma Aldrich Saint-Louis, MO, USA | PKH26GL-1KT | PKH are patented fluorescent dyes and a cell labeling technology named after their discoverer Paul Karl Horan |
| Program for scientific multidimensional images analysis | ImageJ, Research Services Branch, National Institute of Mental Health, Bethesda, Maryland, USA | ||
| RPMI medium 1640 (1x) | Gibco, Paisley, UK | 52400-025 | |
| Sytox green 5 mM Impermeable DNA dye | Gibco, Paisley, UK | 57020 | |
| Trypan blue stain 0,4% | Sigma Aldrich, Germany | 17942E | |
| 96 well, black, flat bottom, tissue culture treated plate | Falcon, NY, USA | 353219 |
References
- Jorgensen, I., Rayamajhi, M., Miao, E. A. Programmed cell death as a defence against infection. Nature Reviews Immunology. 17 (3), 151-164 (2017).
- Rao, A. N., Kazzaz, N. M., Knight, J. S. Do neutrophil extracellular traps contribute to the heightened risk of thrombosis in inflammatory diseases?. World Journal of Cardiology. 7 (12), 829-842 (2015).
- Kessenbrock, K., et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nature Medicine. 15 (6), 623-625 (2009).
- Garcia-Romo, G. S., et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Science Translational Medicine. 3 (73), 73ra20 (2011).
- Lande, R., et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Science Translational Medicine. 3 (73), 73ra19 (2011).
- Meng, H., Yalavarthi, S., et al. In Vivo Role of Neutrophil Extracellular Traps in Antiphospholipid Antibody-Mediated Venous Thrombosis. Arthritis & Rheumatology. 69 (3), 655-667 (2017).
- Desai, J., et al. Particles of different sizes and shapes induce neutrophil necroptosis followed by the release of neutrophil extracellular trap-like chromatin. Scientific Reports – Nature. 7 (1), 15003 (2017).
- Desai, J., et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. European Journal of Immunology. 46 (1), 223-229 (2016).
- Apel, F., Zychlinsky, A., Kenny, E. F. The role of neutrophil extracellular traps in rheumatic diseases. Nature Reviews Rheumatology. , (2018).
- Kraaij, T., et al. A novel method for high-throughput detection and quantification of neutrophil extracellular traps reveals ROS-independent NET release with immune complexes. Autoimmunity Reviews. 15 (6), 577-584 (2016).
- Masuda, S., et al. NETosis markers: Quest for specific, objective, and quantitative markers. Clinica Chimica Acta. 459, 89-93 (2016).
- Brinkmann, V., Goosmann, C., Kuhn, L. I., Zychlinsky, A. Automatic quantification of in vitro NET formation. Frontiers in Immunology. 3, 413 (2012).
- Nakazawa, D., et al. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. Journals of the American Society of Nephrology. 25 (5), 990-997 (2014).
- Konig, M. F., Andrade, F. A Critical Reappraisal of Neutrophil Extracellular Traps and NETosis Mimics Based on Differential Requirements for Protein Citrullination. Frontiers in Immunology. 7, 461 (2016).
- Kenny, E. F., et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. 6, (2017).
- Zhao, W., Fogg, D. K., Kaplan, M. J. A novel image-based quantitative method for the characterization of NETosis. Journal of Immunological Methods. 423, 104-110 (2015).
- Pilsczek, F. H., et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. The Journal of Immunology. 185 (12), 7413-7425 (2010).
- Tanaka, K., et al. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS One. 9 (11), e111888 (2014).
- Fuchs, T. A., et al. Novel cell death program leads to neutrophil extracellular traps. Journal of Cell Biology. 176 (2), 231-241 (2007).
- von Köckritz-Blickwede, M., Chow, O., Nizet, V. Fetal calf serum contains heat-stable nucleases that degrade neutrophil extracellular traps. Blood. 114 (25), 5245-5246 (2009).
- Kraaij, T., et al. Excessive neutrophil extracellular trap formation in ANCA-associated vasculitis is independent of ANCA. Kidney International. , (2018).
- Kraaij, T., et al. The NET-effect of combining rituximab with belimumab in severe systemic lupus erythematosus. Journal of Autoimmunity. , (2018).
- Hahn, J., Schauer, C., et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. The FASEB Journal. (Aug 21), (2018).
