Using Sniper-Cas9 to Minimize Off-target Effects of CRISPR-Cas9 Without the Loss of On-target Activity Via Directed Evolution
Summary
Here, we present a protocol to optimize CRISPR-Cas9 to achieve a higher specificity without the loss of on-target activity. We use a directed evolution approach called Sniper-screen to find a mutant Cas9 with the desired characteristics. Sniper-Cas9 is compatible with truncated single-guide RNAs and delivery in a ribonucleoprotein format, well-known strategies for achieving higher specificities.
Abstract
The development of clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (Cas9) into therapeutic modalities requires the avoidance of its potentially deleterious off-target effects. Several methods have been devised to reduce such effects. Here, we present an Escherichia coli-based directed evolution method called Sniper-screen to obtain a Cas9 variant with optimized specificity and retained on-target activity, called Sniper-Cas9. Using Sniper-screen, positive and negative selection can be performed simultaneously. The screen can also be repeated with other single-guide RNA (sgRNA) sequences to enrich for the true positive hits. By using the CMV-PltetO1 dual promoter to express Cas9 variants, the performance of the pooled library can be quickly checked in mammalian cells. Methods to increase the specificity of Sniper-Cas9 are also described. First, the use of truncated sgRNAs has previously been shown to increase Cas9 specificity. Unlike other engineered Cas9s, Sniper-Cas9 retains a wild-type (WT) level of on-target activity when combined with truncated sgRNAs. Second, the delivery of Sniper-Cas9 in a ribonucleoprotein (RNP) format instead of a plasmid format is possible without affecting its on-target activity.
Introduction
In this paper, we aim to improve the specificity of Cas9 by combining different strategies. Various methods of avoiding the off-target effects of CRISPR-Cas9 have been developed. For example, truncated sgRNAs can be used to achieve higher specificity1. Additionally, the method of Cas9 delivery can be changed from a plasmid format to an RNP format to obtain higher specificity2. Specific amino acid residues of the Streptococcus pyogenes Cas9 (SpCas9) protein have been modified according to the rational design described previously3,4,5. Alternatively, amino acid residues have been altered in a random manner and the Cas9 variants with the highest specificity were identified using either a yeast6 or an E. coli7,8 screening system.
However, many groups have reported that Cas9 variants engineered using the design to debilitate the nonspecific interaction between Cas9 and the substrate exhibit low on-target activities7,8,9,10,11,12. We developed an E. coli-based directed evolution system, Sniper-screen, to screen randomly mutagenized Cas9 variants. An E. coli screening system has advantages over a yeast system because of the faster doubling time and higher transformation efficiencies of E. coli.
Both negative and positive selection, based on three different plasmids and a gene of interest (GOI) integrated into the E. coli genome, are used in Sniper-screen. Cas9 variants are expressed under the CMV-PltetO1 dual-promoter system of a low-copy number plasmid so that candidates identified in E. coli can be tested in mammalian cells without the need for subcloning. The GOI is introduced into the E. coli genome using the Tn7 transposon system. The sgRNA plasmid, which contains a temperature-sensitive origin of replication, expresses an sgRNA targeting the GOI; however, the sgRNA and GOI sequences are not perfectly matched. A perfectly matched sgRNA target site exists on a third plasmid containing the ccdB gene, which encodes a lethal product that poisons gyrase. In this system, cells expressing Cas9 variants with high off-target activities are removed because double-strand breaks (DSBs) are introduced into the mismatched site located in the genomic DNA. On the other hand, cells expressing Cas9 variants with low on-target activities are also removed because of lethal ccdB gene expression. The expression level of the Cas9 variants can be changed by altering the concentration of anhydrotetracycline (ATC), which adjusts the selection force.
We reasoned that locating the mismatched sgRNA target site in the genomic DNA rather than on a plasmid would increase the sensitivity of the system. The advantage of this approach is that there is only one genomic site, whereas there would be many plasmids, each containing a target site, within a single E. coli cell.
Using this system, we identified a Cas9 variant, Sniper-Cas9, which shows WT-level on-target activities and reduced off-target activities compared to WT Cas9. Sniper-Cas9 can achieve even higher specificity ratios by using truncated sgRNAs or RNP-based delivery rather than plasmid-based delivery.
Protocol
1. Integration of a human GOI into the BW25141 E. coli strain
- Cloning of the GOI
- Polymerase chain reaction (PCR) amplify a 500 bp length of a human GOI containing various candidate target sites via standard PCR methods with primers containing NotI and XhoI restriction enzyme sites.
NOTE: In this experiment, the GOI was the human EMX1 gene. - Digest both the PCR product and the pgrg3613 vector with NotI and XhoI restriction enzymes.
- Gel purify the desired fragments (500 bp and 12 kb).
- Ligate the fragments together using T4 ligase. To do this, mix 50 ng of digested pgrg36 and 6 ng of PCR insert in a reaction volume of 20 μL containing 1x ligase buffer and 0.5 U ligase enzyme. Incubate at room temperature (RT) overnight.
- Transform the ligated plasmid into DH5-α competent cells. Grow the resulting transformants on Luria broth (LB) agar plates containing ampicillin (100 μg/mL) at 32 °C and incubate overnight. Pick up a colony and grow it in LB media containing ampicillin overnight. Isolate the plasmid DNA from the E. coli, using a commercially available miniprep Kit. Confirm the insertion of the GOI fragment by Sanger sequencing the plasmid obtained from the mini-plasmid preparation (mini-prep)13.
- Polymerase chain reaction (PCR) amplify a 500 bp length of a human GOI containing various candidate target sites via standard PCR methods with primers containing NotI and XhoI restriction enzyme sites.
- Preparation of the BW25141-GOI
- Transform the obtained pgrg36-GOI plasmid into the BW25141 E. coli strain. It is essential to use a BW25141 strain in order to minimize the number of false positive colonies.
- Grow the transformed cells in the LB buffer at 32 °C overnight. GOI is inserted into the genomic DNA of the BW25141 strain (BW25141-GOI). Remove the pgrg36-GOI plasmid from the BW25141-GOI strain using the standard pgrg36 protocol13. Briefly, dilute a colony (approximately 107-fold) and grow it on an LB plate at 42 °C overnight. Streak the colonies on the LB plate and grow them at 42 °C overnight.
- Confirm the correct GOI insertion by colony PCR, using the primers suggested in the pgrg36 protocol: 5'-GATGCTGGTGGCGAAGCTGT-3' and 5'-GATGACGGTTTGTCACATGGA-3'. The primer amplifies the genomic DNA insertion site, and the size of the resulting PCR product will be 904 bp plus the size of the insert (500 bp in this case).
- Prepare electrocompetent BW25141-GOI cells (a detailed protocol is described in steps 3.2.6–3.2.10).
2. Preparation of the Cas9 variant library
- Library preparation
- Transform the Cas9 vector7 into a commercial E. coli mutator strain (Table of Materials) and follow the manufacturer’s instructions to obtain a variant library (the Mutator library).
- Perform error-prone PCR on the whole WT Cas9 sequence in the Cas9 vector, using an error-prone PCR kit (Table of Materials).
NOTE: The low-error rate protocol was adopted in the Sniper-Cas9 case to avoid disrupting the original function of the protein. - Digest the Cas9 vector with appropriate restriction enzymes. Gel purify the PCR product (from step 2.1.2) and the digested backbone.
NOTE: The size of the SpCas9 gene is about 4.3 kb. XhoI and KpnI were chosen to digest pBLC-SpCas9 vector which was used in the Sniper-Cas9 case. - Assemble the backbone fragment (from step 2.1.3) and the insert amplified using error-prone PCR (from step 2.1.3) via isothermal in vitro recombination.
NOTE: More than 500 ng of backbone is needed to obtain a high concentration of library (error-prone PCR [EP] library). Two different error-prone PCR kits were used to prepare EP libraries in the Sniper-Cas9 case (EP library I and II). - Purify the products from the assembly (from step 2.1.4) using a DNA purification kit that enables low-volume elution (Table of Materials). Elute with 6 μL of nuclease-free water (NFW) and measure the concentration of DNA.
- Transform more than 500 ng of Cas9 library vector (for each of three libraries) into 50 μL of electrocompetent E. coli cells (Table of Materials). See the electroporation protocol in steps 3.2.1-3.2.4. For this library preparation, use 1 mL of SOC medium instead of 250 μL per 50 μL of competent cells.
- Make 1:100, 1:1,000, and 1:10,000 dilutions of the mixture containing the recovered cells with SOC medium. Plate the diluted cells on 100 mm LB agar plates supplemented with chloramphenicol (12.5 μg/mL). Plate the remaining cells on a 245 mm2 plate. Incubate at 37 °C overnight.
- Calculation of library complexity
- Photograph the dilution plates via a gel documentation system or an ordinary digital camera. Run OpenCFU software14 and upload the photographs of the dilution plates. Set the counting area inside a plate and remove false colonies.
- Manually multiply the number of colonies by the dilution factor to obtain the original number of transformants. Convert these numbers into logarithmic form (base 10). Calculate the average to determine the complexity of the library.
- When the desired complexity value is obtained, gather all the colonies on the 245 mm square plate (from step 2.1.7) using a spreader and 20 mL of LB supplemented with chloramphenicol. Do not grow the gathered colonies and purify the plasmid library using a commercial midiprep kit.
NOTE: The higher the library complexity, the better. When Sniper-Cas9 was identified, a diversity of 3 x 106 was achieved for each library.
3. Positive and negative screening for evolving Cas9
- Target selection and plasmid construction
- Select a target sgRNA spacer sequence in the GOI. Substitute one or two residues in the random nucleotide to produce a mismatched sequence.
NOTE: Human EMX1 target site 3 (GAGTCCGAGCAGAAGAAGAA with GGG PAM) was used in the Sniper-Cas9 case. Followings are the mismatched sequences used: GAGTCCGAGCAGAAagAGAA, GAacCCGAGCAGAAGAAGAA, GAGTCCGAGCAGAgGAAGAA, and GAGcCCGAGCAGAAGAAGAA. - Insert the mismatched sequence (see step 3.1.1) into the sgRNA plasmid using standard oligonucleotide (oligo) cloning procedures7.
- Insert the mismatched sequence with a PAM at the 3' end into p11-lacY-wtx1 (Table of Materials) to construct the ccdB plasmid using standard oligo cloning procedures15.
- Select a target sgRNA spacer sequence in the GOI. Substitute one or two residues in the random nucleotide to produce a mismatched sequence.
- Preparation of Sniper-screening E. coli competent cells
- Thaw BW25141-GOI electrocompetent cells on ice.
- Add 1 ng each of the ccdB plasmid and the sgRNA plasmid with double mismatches into 50 μL of thawed BW25141-GOI cells. Gently mix the cells by pipetting and move them into a prechilled 0.1 cm electroporation cuvette.
- Transform the E. coli with the two plasmids via electroporation. Add 250 μL of SOC medium immediately after electroporation. Gently pipette the solution to mix the cells and the medium. Transfer the mixture to a 1.5 mL microcentrifuge tube.
NOTE: For maximum efficiency, set the voltage at 1.80 kV, and the runtime should be between 4.8 ms and 5.0 ms. - Recover the transformed cells and incubate them at 32 °C for 1 h with gentle shaking.
- Plate 125 μL of the recovered cells on an ampicillin (50 μg/mL)/kanamycin (25 μg/mL) LB agar plate (culture condition). Plate the remaining cells on an ampicillin/kanamycin/arabinose (1.5 mg/mL) LB agar plate (ccdB-expressing condition). Incubate at 32 °C overnight.
- Check for the absence of surviving colonies on the ccdB-expressing condition plate. Gather colonies from the culture condition plate using a spreader and culture them in 250 mL of super optimal broth (SOB) medium supplemented with 50 μg/mL of ampicillin and 25 μg/mL of kanamycin at 32 °C, with gentle shaking.
- When the optical density at 600 nm (OD600) reaches 0.4, chill the flask on ice. Prepare prechilled deionized water and a prechilled 10% glycerol solution (sterilize before use).
- Centrifuge (at 4,000 x g for 5 min at 4 °C) the cells and discard the supernatant. Add 200 mL of prechilled deionized water. Resuspend the cells using a 10 mL serological pipette. Repeat this step 3x.
- Wash the cells with 50 mL of prechilled 10% glycerol solution. Centrifuge them as before (at 4,000 x g for 5 min at 4 °C).
- Discard the supernatant and resuspend the pellet in 300 μL of 10% glycerol solution. Make 50 μL aliquots and freeze them in liquid nitrogen. Store the cells (Sniper-screening cells) at -80 °C.
- Sniper-screening
- Transform the Sniper-screening cells (from step 3.2.10) with 100 ng of the Cas9 variant plasmids from each library (from step 2.2.3.See steps 2.1.1 and 2.1.4). Follow the electroporation steps described in steps 3.2.1–3.2.3.
- Transfer 250 μL of the cells to a fresh 1.5 mL microcentrifuge tube. Add 250 pg of ATC to make a final concentration of 10 ng/mL. Recover both ATC-containing and ATC-free cells (see step 3.2.4 for the recovery step).
- Plate 25 μL of recovered ATC-free cells on a chloramphenicol/kanamycin LB agar plate (nonselective condition). Add ATC to the recovered ATC-containing cells to make a final concentration of 100 ng/mL on a 245 mm LB plate. Immediately plate the cells on a chloramphenicol/kanamycin/arabinose LB agar plate (selective condition). Incubate overnight at 32 °C.
NOTE: The size and the number of the LB plate are determined by the size of diversity of the screening covers. In the case of a 100 mm Petri dish with 20 mL of LB, add 2 μg of ATC. - Photograph the plates. Count the number of viable colonies using OpenCFU software14. (See step 2.2.1) Make sure that the number of colonies on the nonselective plate is at least 10x larger than the diversity of the library to cover all variants.
- Calculate the survival frequency as follows.
Survival frequency = the number of colonies on a selective plate / (the number of colonies on a nonselective plate x 10) - Pool the colonies that survived on the selective plates from all three libraries. Incubate the surviving colonies in 250 mL of LB medium supplemented with 12.5 μg/mL chloramphenicol at 42 °C overnight. Isolate the screened Cas9 library DNA using a midiprep kit.
NOTE: This step clears the sgRNA plasmid. - Repeat the screening process from steps 3.3.1–3.3.6 until the survival frequency reaches a plateau. Use 10 ng of the selected Cas9 plasmid for transformation and 10 ng/mL of ATC during recovery. Maintain the ATC concentration at 100 ng/mL for the selective condition.
- Shuffling and the second screening
- Shuffle the selected pooled variants using the following DNA-shuffling protocol. PCR amplify the Cas9 insert in the Cas9 plasmid using flanking primers, 150 nucleotides from the insert boundaries. Digest 2 μg of amplified PCR product with DNase I for 1 min at 37 °C.
- Purify fragments 70–200 bp in length using 2% agarose gel electrophoresis. PCR amplify the purified fragments. Use the product as a template to PCR amplify the Cas9 insert with appropriate primers flanking Cas9. Use the final PCR product to construct a Cas9 library as described in step 2.1.4.
- Prepare new Sniper-screening cells (see section 3.2) with another mismatched sgRNA plasmid (see step 3.1.1). Redo the screening process (sections 3.2–3.3) until the survival rate reaches a plateau. Use 10 ng of the selected Cas9 plasmid for transformation and 10 ng/mL ATC during recovery. Maintain the ATC concentration at 10 ng/mL for the selective condition.
- Selection of evolved Cas9 mutant plasmids
- After the last screening step, randomly pick one hundred colonies from the selective plate and culture them in chloramphenicol-containing LB medium at 42 °C overnight.
- Isolate the plasmids using a miniprep procedure and Sanger-sequence the inserts using sequencing primers within Cas9.
- Select the top three most frequent variants to test them in human cell lines.
4. Delivery of Cas9 as an RNP with truncated sgRNA
- Target gene selection using Cas-OFFinder
- Pick target sites with Cas-OFFinder (http://www.rgenome.net/cas-offinder/). Select the PAM type appropriate for the specific type of Cas9 and the target genome (human, mouse, zebrafish, etc.). Fill in the Query sequences tab, choose the Mismatch number, and click the Soumettre button.
- After a few seconds, the on-target (with a mismatch number of ‘0’) and off-target sites will appear. In general, choose the off-target sites with one to three mismatches.
- Preparation of the sgRNA template
- Order crRNA and tracrRNA oligos with the following template sequences, namely crRNA sequence: 5'-TAATACGACTCACTATAGGNNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAA-3'; tracrRNA sequence: 5'-AGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3'. Fill N20 in the crRNA sequence with the target sequence obtained in step 4.1.2. To design truncated sgRNAs, remove bases from the 5' end to obtain N19, N18, or N17 sgRNA sequences.
- Prepare the PCR mixture for amplification of the sgRNA-encoding sequence as follows: combine 10 μL of 5x buffer, 0.5 μL of crRNA oligo (100 pmol/μL), 0.5 μL of tracrRNA oligo (100 pmol/μL), 2.5 μL of dNTPs (10 mM each), 0.5 μL of DNA polymerase, and 36 μL of NFW (Table of Materials).
- Amplify the template using the following conditions, namely, for the initial denaturation: 1 min at 98 °C; for the denaturation, annealing, and extension: 10 s at 98 °C, 15 s at 54 °C, 20 s at 72 °C, for 25 cycles; for the final extension: 5 min at 72 °C.
- Analyze 2 μL of the amplified template DNA (from step 4.2.3) on a 2% agarose gel. Purify the template using a PCR purification kit.
NOTE: The size of the template DNA is 125 bp.
- Synthesis of sgRNA
- Prepare the reaction mixture for sgRNA synthesis as follows: combine 8.5 μL of template DNA (from step 4.2.3), 1 μL of UTP (25 mM), 1 μL of CTP (25 mM), 1 μL of GTP (25 mM), 1 μL of ATP (25 mM), 4.2 μL of MgCl2 (100 mM), 4.5 μL of T7 RNA polymerase (50 U/μL), 3 μL of 10x T7 RNA polymerase buffer, 1.2 μL of pyrophosphatase (0.5 U/μL), 0.75 μL of RNase inhibitor (40 U/μL), and 4.2 μL of NFW.
- Incubate the reaction mixture at 37 °C overnight (at least for 10 h). Add 0.5 μL of DNase (2 U/μL) to the reaction mixture and incubate at 37 °C for 15–30 min. Purify the sgRNA using an RNA purification kit.
- To reduce the toxicity caused by an innate immune response triggered by the 5'-triphosphate on the sgRNA16, remove the 5'-triphosphate from the guide RNAs with calf intestinal alkaline phosphatase (CIP) as follows: treat 10 µg of in vitro-transcribed RNA with 250 U of CIP for 3 h at 37 °C in the presence of 100 U of RNase inhibitor. Purify the CIP-treated sgRNA using an RNA purification kit.
5. WT- and Sniper-Cas9 protein expression and purification
- Protein expression in E. coli
- Transform pET plasmids18 encoding His-tagged WT- and Sniper-Cas9 into the BL21 (DE3) E. coli strain.
- Inoculate 50 mL of LB medium containing 50 μg/mL kanamycin with a fresh colony harboring the pET-Cas9 expression plasmid and shake it overnight (200 rpm) at 37 °C (preculture).
- Transfer 10 mL of overnight culture to 500 mL of fresh LB medium containing 50 μg/mL kanamycin. Incubate the culture with shaking (200 rpm) at 37 °C for 2 h.
- Monitor the OD600 until the culture reaches the mid-log phase of growth (OD600 ≈ 0.6–0.7).
- Induce the expression of the WT- or Sniper-Cas9 protein with isopropyl β-D-1-thiogalactopyranoside (IPTG, at a final concentration of 0.25 nM). Incubate the culture at 18 °C overnight.
- Protein purification
- Harvest the cells by centrifugation at 5,000 x g for 10 min at 4 °C.
- Resuspend the pellet in the lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 4 mM dithiothreitol [DTT], 5 mM benzamidine, 100 mM phenylmethylsulfonyl fluoride [PMSF], pH 8) at 20 mL per gram wet weight.
- Add PMSF, DTT, and lysozyme to a final concentration of 1 mg/mL each and incubate the mixture on ice for 30 min.
- Sonicate the cells on ice. Pulse repeatedly for 10 s at 200–300 W with a 10 s cooling period between each pulse, for a total time of 20 min.
- Centrifuge the lysate at 6,000 x g for 30 min at 4 °C.
- Remove the supernatant to a fresh tube. Add 1 mL of His-Bind agarose resin to 5 mL of cleared lysate and shake gently for 1 h at 4 °C.
- Load the lysate/His-Bind Agarose resin mixture onto a column with a capped bottom outlet.
- Remove the bottom cap and collect the column flow-through.
- Wash the column 2x with wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8); collect the wash fraction for analysis by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
- Elute the protein 10x with 1 mL of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8), collecting samples for SDS-PAGE.
- Concentrate the eluted WT- or Sniper-Cas9 protein using a 100 kDa column filter. Store the samples in a solution of 10 mM Tris-HCl, 150 mM NaCl, and 50% glycerol at -80 °C.
6. RNP delivery
- Transfection and preparation of cells for RNP delivery
- Maintain HEK293T cells in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics at 37 °C with 5% CO2.
- Mix WT- or Sniper-Cas9 protein (2 μg) with sgRNA (2 μg) and incubate for 10 min at RT to make RNP complexes.
- Trypsinize and count the cells. Prepare 2 x 104 cells per one reaction. Wash the cells with phosphate-buffered saline (PBS) and centrifuge. Aspirate the supernatant and resuspend the pellet with electroporation buffer.
- Electroporate RNP complexes into the cells using the following settings, namely 1,300 V, 30 ms, and one pulse. Plate the cells on a 48-well plate filled with 500 μL of DMEM supplemented with FBS and antibiotics (as described in step 6.1.1) right after the electroporation. Incubate at 37 °C with 5% CO2.
- Isolate the genomic DNA with a gDNA preparation kit, 48 h after the transfection.
7. Transfection of plasmids encoding Sniper-Cas9 and sgRNA
- Construction of an sgRNA plasmid
- Order forward and reverse oligos with the following template sequences, namely forward: 5'-CACCGNNNNNNNNNNNNNNNNNNNN-3'; reverse: 5'-AAACNNNNNNNNNNNNNNNNNNNNC-3'. Replace N20 with the target sequence obtained in step 4.1.2. Shorten the target sequence to a length of N19, N18, or N17 to synthesize truncated sgRNA.
- Anneal both oligos in 1x T4 DNA ligase buffer.
- Digest the pRG2 vector with BsaI restriction enzyme.
- Gel purify the digested vector (3,300 bp) using a 0.8% agarose gel.
- Ligate the annealed oligo and the purified fragment using T4 ligase at 37 °C: mix 50 ng of digested pRG2 and 1 ng of annealed oligo in a reaction volume of 20 μL. Incubate at RT for 15 min.
- Transform the ligation mixture into the DH5 alpha strain and grow transformants on LB agar plates containing ampicillin (100 μg/mL) at 37 °C. Confirm the insertion of the oligo in the vector by standard sequencing.
- Transfection of plasmids encoding Sniper-Cas9 and sgRNA
- Maintain HEK293T cells in DMEM supplemented with 10% FBS and 1% antibiotics at 37 °C with 5% CO2. The day before transfection, trypsinize and count the cells. When working at a 48-well scale, plate 1 x 105 cells per well in 250 μL of complete growth medium. The cells should be 50%–80 % confluent on the day of transfection.
- At the 48-well scale, prepare 250 ng of p3s-Cas9 plasmid and 250 ng of sgRNA plasmid for transfection, using a lipid-based transfection reagent. Mix the plasmids in 25 μL of serum free-MEM.
- Dilute 1 μL of transfection reagent with 25 μL of serum free-MEM. Incubate the mixture at RT for 5 min. Combine the two mixtures and incubate the resulting solution at RT for 20 min to form plasmid-lipofectamine complexes.
- After 20 min of incubation, add 50 μL of the solution containing the plasmid-transfection reagent complexes directly to each well containing cells, and mix gently by rocking the plate back and forth. Incubate the cells at 37 °C in a CO2 incubator for 48–72 h post transfection before assaying for transgene expression.
8. Calculation of indel frequencies to determine on-target and off-target activities
- Targeted deep sequencing for the analysis of on-target and potential off-target sites
- Isolate genomic DNA from step 6.1.5 or 7.2.4 with a gDNA preparation kit. Generate deep sequencing libraries by PCR amplification of the gDNA with primers targeting on-target and off-target.
- Use index primers to label each sample. Subject pooled libraries to paired-end sequencing using a next-generation sequencing machine.
- Deep sequencing analysis using Cas-Analyzer
- Analyze deep sequencing data using the Cas-Analyzer assessment tool17.
- Choose the Fastq files under the Read 1 and Read 2 tabs (Read 1 = XX_SXX_L001_R1_001.fastq, Read 2 = XX_SXX_L001_R2_001.fastq).
- Fill out the Basic Information tab and the Analysis Parameters tab. Click the Soumettre button.
Representative Results
After Sniper-screen is performed, the percentage of survival colonies can be calculated by dividing the number of colonies on the LB plate containing chloramphenicol, kanamycin, arabinose, and ATC (CKAA) by the number of colonies in the LB plate containing chloramphenicol and kanamycin only (CK). This percentage was usually very low when Sniper-screen was performed with the libraries of SpCas9. True-positive hits can be enriched by repeating the screen with the surviving pool. In this representative Sniper-screen, for example, a 100% survival rate was obtained after the third screen (Figure 1). Transfections using RNPs or plasmid-encoded Sniper-Cas9 can be done for various targets and the resulting on-target and off-target activities measured by targeted amplicon sequencing (Figure 2). At most targets, Sniper-Cas9 shows the same level of on-target activities and higher specificity ratios compared to the WT. Truncated sgRNAs can also be used to further improve specificity (Figure 3). However, their use is limited to only a few targets because they result in low on-target activities compared to full-length sgRNAs, in most cases. Therefore, sgRNAs with varying lengths (from 17- to 20-mers) must be tested and both on-target and off-target activities must be measured to optimize specificity.
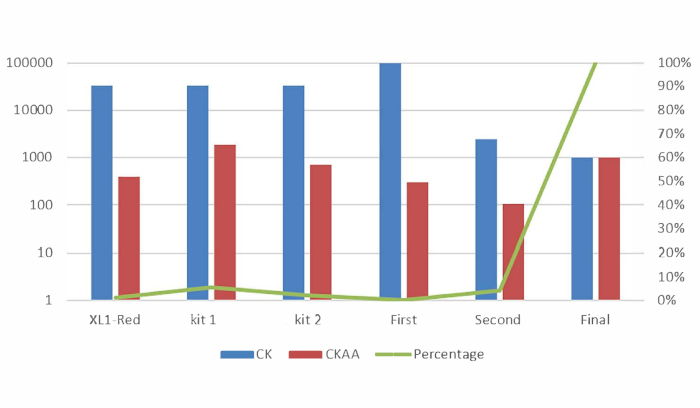
Figure 1: Representative Sniper-screens performed with different libraries of random Cas9 variants. Mutator library, EP library I, and EP library II indicate libraries made using different commercial kits. First, second, and final indicate the number of times the enrichment screen was performed7. This figure has been modified from Lee et al.7. Please click here to view a larger version of this figure.
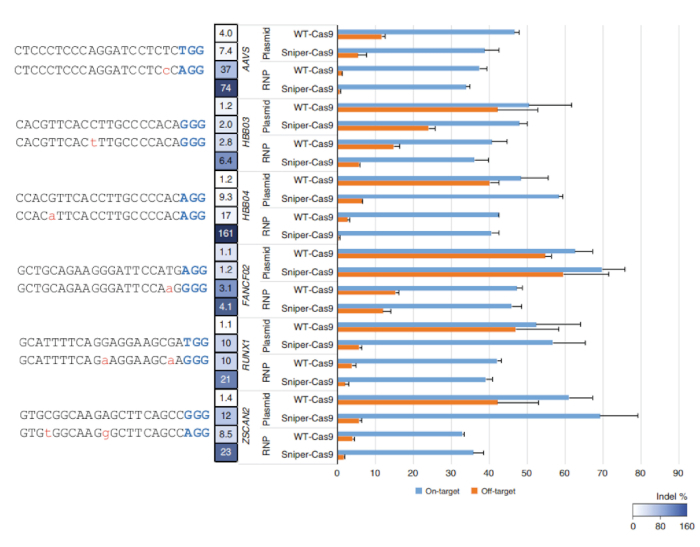
Figure 2: On-target and off-target activities of WT-Cas9 or Sniper-Cas9 paired with a 20-mer guide sequence delivered via plasmid or RNP. Specificity ratios were determined by dividing the on-target activity by the off-target activity. Error bars indicate SEM (n = 3)7. This figure has been modified from Lee et al.7. Please click here to view a larger version of this figure.
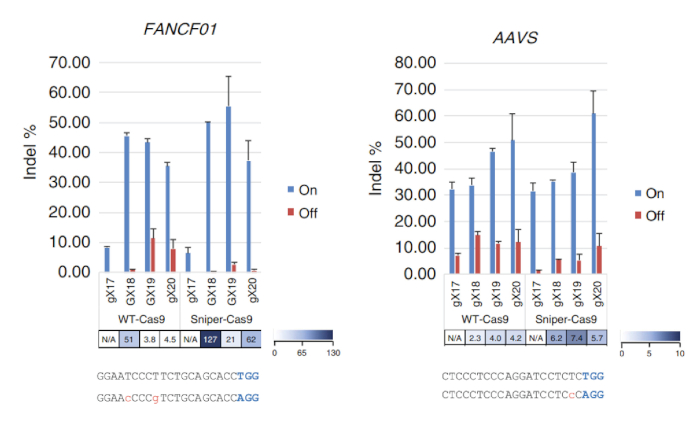
Figure 3: On-target and off-target activities of Sniper-Cas9 compared to WT-Cas9 obtained using sgRNAs with variable lengths targeting the FANCF01 and AAVS sites. Specificity ratios were determined by dividing indel frequencies at on-target sites by those at the respective off-target sites. sgRNAs with a matched guanine at the 5' terminus (GX18 or GX19) and those with a mismatched guanine (gX17, gX18, gX19, or gX20) are indicated. Specificity ratios were not calculated when the normalized on-target activities were <70%7. This figure has been modified from Lee et al.7. Please click here to view a larger version of this figure.
Discussion
For those who want to avoid cumbersome screening procedures to obtain Sniper-Cas9, the Sniper-Cas9 protein and the encoding plasmid are available. Using these materials, the optimum length of the sgRNA, that providing the highest specificity ratio, should be determined. In addition, delivery of Sniper-Cas9 and sgRNA in an RNP format is recommended because it usually results in a higher specificity ratio than delivery in a plasmid format. Unlike Sniper-Cas9, other engineered Cas9 variants are not compatible with truncated sgRNAs6,7 or delivery in RNP form8 (with the exception of HiFi-Cas9).
For Sniper-screen, the selection of the mismatched sequence is the most important step. Selection of a mismatched sgRNA that is associated with low cleavage activity toward the GOI sequence should be avoided. If not, Cas9 variants with a WT level of specificity will not cleave the E. coli genomic DNA with the mismatched target site. This effect will result in a large number of background colonies, jeopardizing the whole screening procedure.
Because E. coli has a fast doubling time and high transformation efficiency compared to yeast, Sniper-screen is advantageous compared to yeast-based screening methods. Additionally, Sniper-screen should be more sensitive than other E. coli-based systems in which the mismatched site is carried on a plasmid: there is one copy of the genomic DNA and thus only one copy of the mismatched site in our system, but a large number of plasmids within a single E. coli cell.
The specificities of other DNA endonucleases that induce DSBs, such as SaCas9 or Cpf1s, could also be improved by using Sniper-screen. Unfortunately, Sniper-screen cannot be used to increase the specificity of base editors directly, because base editors do not induce DSBs in the genomic DNA of E. coli. As base editors use the nickase or dead version of Cas9 in the core of their system, the specificities of base editors could be increased by using the hits obtained from Sniper-screen.
Divulgations
The authors have nothing to disclose.
Acknowledgements
This research was supported by grants from the Ministry of Science and ICT of Korea (2017M3A9B4061406) and the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (grant numbers 2017M3A9B4061404 and 2018M3A9H3020844) to Jungjoon K. Lee. The plasmid encoding pGRG36 was a gift from Nancy Craig (Addgene plasmid #16666) and p11-LacY-wtx1 was a gift from Huimin Zhao.
Materials
| Alkaline Phosphatase, Calf Intestinal (CIP) | NEB | M0290L | |
| Ampicillin Sodium Salt | Fisher Chemical | BP1760-25 | |
| Anhydrotetracycline hydrochloride | Sigma Aldrich | 37919 or 94664 | |
| Antibiotic Antimycotic solution | Welgene | LS203-01 | Media component |
| BamHI | Enzynomics | R003L | |
| Chloramphenicol | Sigma Aldrich | R4408 | |
| Diversify PCR Random Mutagenesis | Clontech | 630703 | Error-prone PCR kit |
| DNA-Shuffling Kit | Jena Bioscience | PP-103 | |
| Dulbecco's Modified Eagle's Medium (DMEM) | Welgene | LM001-17 | Culture media |
| Endura Electrocompetent Cells (DUO) | Lucigen | 60242-2 | E. coli electrocompetent cells for library preparation |
| Fetal Bovine Serum (FBS) | Welgene | S101-01 | Media component |
| Gene Pulser II | Bio-Rad | E. coli electroporation equipment | |
| GeneMorph II Random Mutagenesis Kit | Agilent | 200550 | Error-prone PCR kit |
| genomic DNA prep kit | GenAll | 106-152 | gDNA-prep kit |
| Glycerol | BioShop | GLY001.1 | |
| GP/MP Cuvette, 0.1cm | Bio-Rad | BR165-2089 | E. coli electroporation equipment |
| HEK293T cell | ATCC | CRL-11268 | Human cell line |
| Kanamycin sulfate | Acros Organics | 450810500 | |
| L-(+)-Arabinose | Sigma Aldrich | A3256-25G | |
| LaboPass PCR Purification Kit | Cosmogenetech | CMR0112 | |
| LaboPass Plasmid Mini | Cosmogenetech | CMP0112 | Mini-prep kit |
| LB Agar Miller | Formedium | LMM0202 | |
| LB Broth Miller | Formedium | LMM0102 | |
| Lipofectamine | Invitrogen | 11668019 | Transfection reagent |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | E2621L | Gibson assembly (isothermal in vitro recombination) mixture |
| Neon Transfection System | Thermo | MPK5000 | RNP Electroporation equipment |
| Neon Transfection System 10 µL Kit | Thermo | MPK1096 | RNP Electroporation equipment |
| NucleoBond Xtra Midi EF | Macherey-Nagel | 740420.50 | Midi-prep kit |
| Oligo Clean & Concentrator | Zymo Research | D4061 | DNA purification kit that enables low-volume elution |
| Opti-MEM | Gibco | LS31985070 | Transfection media |
| p11-lacY-wtx1 | Addgene | #123945 | |
| pgrg36 | Addgene | #16666 | E. coli mutator strain |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530L | |
| Pyrophophatase | NEB | M2403L | |
| Ribonucleotide Solution Set | NEB | N0450L | |
| RNase inhibitor | NEB | M0314L | |
| T7 RNA polymerase | NEB | M0251L | |
| Turbo Dnase | Ambion | AM2238 | |
| XbaI | Enzynomics | R013L | |
| XL1-Red Competent Cells | Agilent | 200129 |
References
- Fu, Y., Sander, J. D., Reyon, D., Cascio, V. M., Joung, J. K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nature Biotechnology. 32 (3), 279-284 (2014).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Chen, J. S., et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 550 (7676), 407-410 (2017).
- Kleinstiver, B. P., et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 529 (7587), 490-495 (2016).
- Slaymaker, I. M., et al. Rationally engineered Cas9 nucleases with improved specificity. Science. 351 (6268), 84-88 (2016).
- Casini, A., et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nature Biotechnology. 36 (3), 265-271 (2018).
- Lee, J. K., et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nature Communications. 9 (1), 3048 (2018).
- Vakulskas, C. A., et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nature Medicine. 24 (8), 1216-1224 (2018).
- Anderson, K. R., et al. CRISPR off-target analysis in genetically engineered rats and mice. Nature Methods. 15, 512-514 (2018).
- Kim, S., Bae, T., Hwang, J., Kim, J. S. Rescue of high-specificity Cas9 variants using sgRNAs with matched 5′ nucleotides. Genome Biology. 18 (1), 218 (2017).
- Kulcsar, P. I., et al. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biology. 18 (1), 190 (2017).
- Zhang, D., et al. Perfectly matched 20-nucleotide guide RNA sequences enable robust genome editing using high-fidelity SpCas9 nucleases. Genome Biology. 18 (1), 191 (2017).
- McKenzie, G. J., Craig, N. L. Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiology. 6, 39 (2006).
- Geissmann, Q. OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS One. 8 (2), e54072 (2013).
- Chen, Z., Zhao, H. A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Research. 33 (18), e154 (2005).
- Kim, S., et al. CRISPR RNAs trigger innate immune responses in human cells. Genome Research. 28 (3), 367-373 (2018).
- Park, J., et al. Cas-Analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics. 33, 286-288 (2017).
- Studier, F. W., et al. Use of T7 RNA Polymerase to Direct Expression of Cloned Genes. Methods in Enzymology. 185, 60-89 (1990).
