Table 1 shows the expected cell yields and viability from adult mouse ear (Figure 1) and neonatal skin under non-pathological conditions. The table also shows representative data of animals from a mixed C57/126 background. It is expected that the skin of other strains would result in similar cell yields and viabilities. The approximate yield is dependent on the surface area of skin and indicates that neonatal skin would be a better choice for experiments that require larger numbers of cells (Table 1). Low yields or reduced viability (<50%) would indicate issues with digestion or experimental conditions that reduce skin integrity or induce of cell death. Culturing cells with appropriate media and supplements can help assess the quality of KC preparations17.
Following normalization20 and deconvulation18 of FCS files, gating of data will define epidermal cell populations of interest and demark individual cell cycle phases (Figure 2). In bivariate plots comparing the 191Ir vs 193Ir channels, intact cells can be gated in the upper right quadrant (Figure 2A). Plotting event length vs 191Ir and selecting for a tight cluster of cells near the 191Ir axis demarks single cells. Viable cells are selected in a plot of 195Pt vs 193Ir plot by gating for cells with low 195Pt levels. The sample shown in Figure 2A has debris and increased presence of cisplatin labeled non-viable cells. This may indicate that the sample was over-digested, harshly handled, or left on ice or RT for too long before staining for mass cytometry. Profiles from tissue-cultured cells typically give higher percentages of 195Pt negative cells (Figure 2B). Alternatively, the presence of 195Pt positive cells may be expected if the induction cell death is a feature of the experimental model and/or conditions.
Ideally, markers that define the population of interest should be included in the analysis of specific cell types. For example, immune cells that are expected to be present in crude KC suspensions can be detected by the addition of a CD4523 antibody, which detects hematopoietic cells (Figure 2C). With regards to the proliferation antibody panel13, plotting IdU vs CYCLIN B1 resolves cell cycle phases (Figure 2C) similar to standard BrdU/PI flow plot (Figure 2D), allowing for the detection of S-phase, G0/G1 and G2/M cell populations. In IdU vs pHH3 plots, high pHH3 positivity identifies M phase cells. Lastly, gating G0/G1 cells on high pRB signal can distinguish G0 (quiescent) from cells in G1 (Figure 2C, left most panel).
Having identified the different cell cycle phases, it is then possible to construct a graphical representation of cell cycle profiles for different cell types or experimental conditions. For example, distinct cell cycle profiles emerge when comparing CD45- vs CD45+ cell populations (Figure 3A) in the KC suspension shown in Figure 2. As expected hyperplastic KCs found in the CD45- group had fewer cells in G0 and more cells in S, G2, and M phases3. In contrast, immune cells from the same sample had notable populations in G0 and G1. In addition to cell cycle profiles, the characterization of gene expression in a cell cycle-dependent manner is also possible. For example, phospho proteins that help define EGFR and mTOR/PI3K signaling were analyzed in the data presented for Figure 3A. Analysis of the G1 phases of CD45- vs CD45+ cells revealed a similar profile for EGFR and mTOR/PI3K signaling proteins, although there appeared to be slight differences in the percentage of pS6 and pEGFR positive cells (Figure 3B). Similar approaches can be adapted to characterize the expression of other signaling pathway proteins or cellular processes at different phases of the cell cycle.
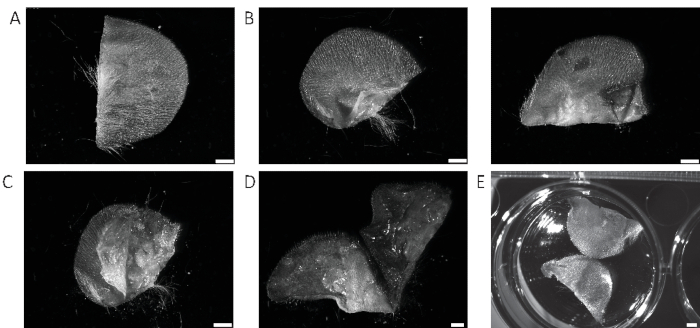
Figure 1: Preparation of mouse ear skin for digestion. (A) First, remove the ear from the animal and lay flat on a clean Petri dish. (B) Grasp one side of the ear at either the middle or edge using curved precision forceps. (C) Flip over and using another pair of forceps and gently pull the halves of the ear apart until the ear tears on the crease. (D) Continue pulling until the sides separate into two halves. (E) Float the ear halves on dissociation media with the dermis side touching the solution. Scale bar = 2 mm. Please click here to view a larger version of this figure.
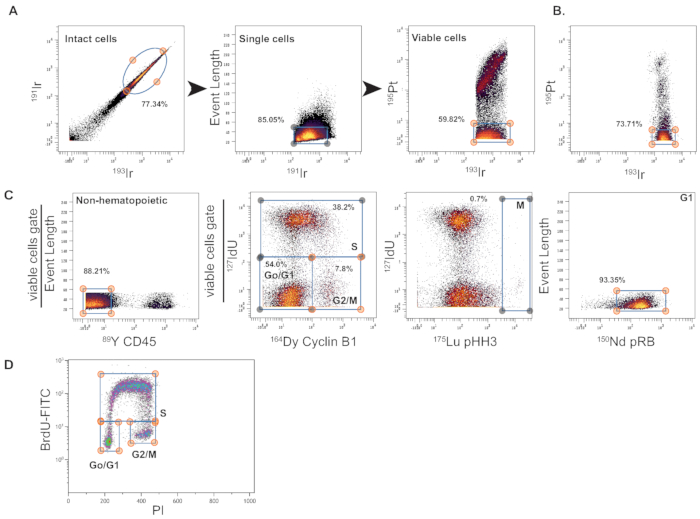
Figure 2: Gating strategy for mass cytometry data. The data in this figure was generated from an experimental animal treated with the phorbol ester TPA as previously described3. TPA is a PKC activator that induces skin inflammation24. (A) The panels show the initial gating strategy after normalization and deconvolution of barcoded data. By gating on event length, 191Ir,193Ir, and 195Pt channels, it is possible to select for intact, single, and viable cells. (B) Human SCC SRB-P93 cells treated with DMSO and were then stained for mass cytometry. Data was gated as in (A) and the plot shows the levels of live cells in this sample. (C) The inclusion of lineage markers allows for the selection of cell populations of interest. For this example, viable cells from (A) were gated on event length vs. CD45 to exclude hematopoietic cells. Gating CD45- cells for incorporation of IdU vs CYCLIN B1 allows the identification of S, G0/G1, and G2/M cell populations. Selection of pHH3 high cells defines M phase, whereas the analysis of G0/G1 for pRB positivity identifies cells in G1. (D) The plot shows the representative results of cell cycle analysis by the detection of BrdU incorporation and PI (DNA content) with fluorescence-based flow cytometry. The data shown is from human Colo163 SCC cells grown with BrdU for 1 h as previously described3. Please click here to view a larger version of this figure.
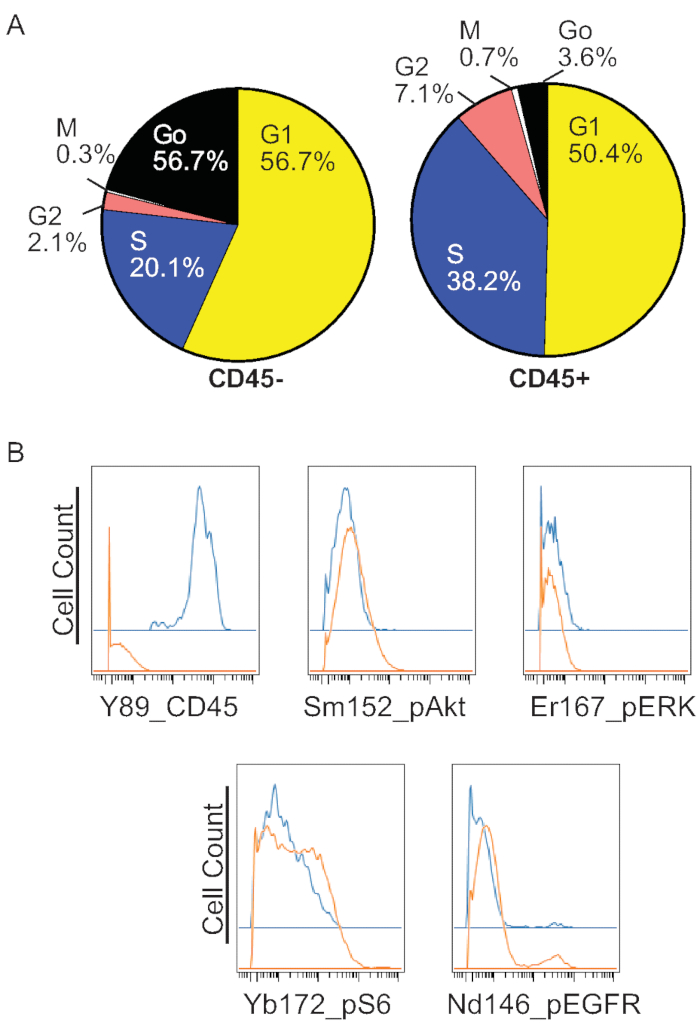
Figure 3: Characterization of cell cycle profiles and phase-specific protein expression. (A) The cell cycle profiles were determined in the sample from Figure 2 for CD45- vs CD45+ cells. (B) Analysis of the G1 phases with phospho-specific antibodies in CD45-(orange) and CD45+(Blue) cells revealed similar expression profiles for markers of the EGFR and mTOR/PI3K signaling pathways. Please click here to view a larger version of this figure.
| Tissue | Area | Cell yield | Viability |
| Mouse ear | 225-230 mm3 | 1-2×106 | 70-90% |
| Neonatal skin | 1000-1100 mm3 | 5-10×106 | 80-99% |
Table 1: Typical Cell yields and viability from adult mouse ear and neonatal skin. Cell counts and viability by Trypan blue exclusion were averaged from ear KCs harvested from n=7 8-10 week old adult mouse ears or n=7 P3 neonatal skins.
