Robust Comparison of Protein Levels Across Tissues and Throughout Development Using Standardized Quantitative Western Blotting
Summary
This method describes a robust and reproducible approach for the comparison of protein levels in different tissues and at different developmental timepoints using a standardized quantitative western blotting approach.
Abstract
Western blotting is a technique that is commonly used to detect and quantify protein expression. Over the years, this technique has led to many advances in both basic and clinical research. However, as with many similar experimental techniques, the outcome of Western blot analyses is easily influenced by choices made in the design and execution of the experiment. Specific housekeeping proteins have traditionally been used to normalize protein levels for quantification, however, these have a number of limitations and have therefore been increasingly criticized over the past few years. Here, we describe a detailed protocol that we have developed to allow us to undertake complex comparisons of protein expression variation across different tissues, mouse models (including disease models), and developmental timepoints. By using a fluorescent total protein stain and introducing the use of an internal loading standard, it is possible to overcome existing limitations in the number of samples that can be compared within experiments and systematically compare protein levels across a range of experimental conditions. This approach expands the use of traditional western blot techniques, thereby allowing researchers to better explore protein expression across different tissues and samples.
Introduction
Western blotting is a technique that is commonly used to detect and quantify protein expression, including in tissue homogenates or extracts. Over the years, this technique has led to many advances in both basic and clinical research, where it can be used as a diagnostic tool to identify the presence of disease1,2. Western blotting was first described in 1979 as a method to transfer proteins from polyacrylamide gels to nitrocellulose sheets and subsequently visualize proteins using secondary antibodies that were either radioactively labelled or conjugated to fluorescein or peroxidase3. Through the development of commercially available kits and equipment, Western blotting methods have been increasingly standardized and simplified over the years. Indeed, the technique is now readily performed by scientists with varying backgrounds and levels of experience. However, as with many similar experimental techniques, the outcome of Western blot analyses is easily influenced by choices made in the design and execution of the experiment. It is important, therefore, that the accessibility of standardized Western blotting methods does not obscure the need for careful experimental planning and design. Experimental considerations include, but are not limited to, sample preparation and handling, selection and validation of antibodies for protein detection, and gel-to-membrane transfer efficiency of particularly small or large (<10 or >140 kDa) proteins4,5,6,7,8,9. Protein quality of the original sample plays a significant role in determining the outcome of the subsequent Western blot analysis. As protein can be extracted from a wide variety of samples and sources, including cell lines, tissues from animal models, and post-mortem human tissues, consistency in handling and processing is required to obtain reproducible results. For example, when long-term storage of samples for protein extraction is required, it is important to realize that, although protein is generally stable at -80 °C, differences in protein stability between extracted proteins and intact tissues at -80 °C have been reported10. Moreover, to obtain reproducible estimates of protein quantities, consistent homogenization of samples is crucial. Optimizing different lysis buffers and homogenization methods (e.g., manual homogenization compared to automated methods) may be required before starting a large-scale quantitative experiment.
Normalization strategies to correct for protein loading and quantification variability are essential to obtain robust, quantitative results of protein expression. Housekeeping proteins such as β-actin, α-tubulin, β-tubulin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) have traditionally been used to normalize protein levels for quantification. However, normalization to specific housekeeping proteins for quantification purposes has been increasingly criticized over the past few years11,12. For example, the expression of housekeeping proteins can change across different developmental stages13,14, across tissues from the same animal4, and under various disease conditions4,15,16,17. Therefore, the use of specific housekeeping proteins limits the possibilities of making more complex comparisons between protein expression from different tissues, at different timepoints and under varying experimental conditions. An alternative to housekeeping proteins to control for protein loading variation is the use of a total protein stain (TPS) that labels and visualizes all proteins present in a sample. TPS allows signal normalization based on total protein load rather than levels of one specific protein and therefore quantification of TPS signal should be comparable and reproducible regardless of experimental condition, sample type or developmental timepoint. Examples of total protein stains include Ponceau S, stain-free gels, Coomassie R-350, Sypro-Ruby, Epicocconone, Amydo Black, and Cy5 (reviewed in ref. 18). Each of these methods has specific advantages and limitations and method selection depends on the time and tools available as well as the experimental setup4,18.
In addition to using a TPS to correct for within-membrane loading and quantification variability, it may be necessary to compare samples between different membranes, particularly when performing large-scale protein expression analysis. However, variability in factors such as antibody binding efficiency and total protein stain intensity may introduce further variability between protein samples that are analyzed on separate gels and membranes. For robust quantification in this situation, it is therefore necessary to introduce a further normalization step to account for between-membrane variability. This can be achieved by including an internal loading standard on each of the separately analyzed membranes that is kept constant across experiments. This standard can take the form of any protein lysate that can be obtained in sufficient quantities to be used across all membranes included in the experiment. Here, we use a lysate of mouse brain (obtained from 5 day old control mice), as brain is readily homogenized and the obtained protein lysate contains a significant amount of protein at a high concentration. Loading an internal standard in triplicate allows samples on separate membranes to be normalized and compared directly.
Here, we describe a detailed protocol that we have developed to allow us to undertake complex comparisons of protein expression variation across different tissues, mouse models (including disease models), and developmental timepoints19. By combining a fluorescent TPS with the use of an internal loading standard, we were able to overcome existing limitations in the number of samples and experimental conditions that can be compared within a single experiment. This approach expands the use of traditional Western blot techniques, thereby allowing researchers to better explore protein expression across different tissues and samples.
Protocol
Tissues for this procedure were obtained from animal studies that were approved by the internal ethics committee at the University of Edinburgh and were performed in concordance with institutional and UK Home Office regulations under the authority of relevant personal and project licenses.
NOTE: This protocol has been optimized using standardized, commercially available kits and reagents in order to increase reproducibility (see Table of Materials).
1. Preparation of samples
- Protein extraction
- Transfer snap-frozen cell or tissue samples from -80 °C on dry ice, thaw on ice, and wash as required with ice cold 1x phosphate-buffered saline (PBS) (for details on tissues and PBS washes, see Table 1). Avoid unnecessary freeze-thaw cycles as this will affect protein quality.
- Add radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing 1x protease inhibitor to each sample, using the optimal amount per tissue weight (see Table 1 for recommendations).
NOTE: Depending on the application, the type and amount of homogenization buffer may need further optimization. - Use a hand-held electric homogenizer with a polypropylene pestle to homogenize tissue samples. Between each sample, wash the pestle in double-distilled water and dry with a clean tissue. Change the pestle between different experimental conditions and tissues.
- Leave the samples on ice for 10 min after homogenization. Centrifuge the samples at >10,000 x g at 4 °C for 10 min.
- Transfer the supernatant to a new tube on ice without disturbing the pellet. The supernatant is the protein sample. Store the extracted protein at -80 °C or directly proceed to measuring the protein concentration.
- Quantification and normalization of protein concentration
- Measure the protein concentration using a bicinchoninic acid (BCA) assay. Prepare a BCA assay mix according to the manufacturer’s instructions in a 96-well optical plate using 200 µL of BCA mix per well.
NOTE: Other quantification methods such as Lowry and Bradford assays can also be used to determine protein concentration as long as protein concentration is quantified consistently across experiments. - Prepare bovine serum albumin (BSA) standards at increasing concentrations in triplicate and add 1 µL of each protein sample in duplicate. Incubate the 96-well plate in a preheated heat block at 60 °C for 10 min or longer if the protein concentration is expected to be low.
- After incubation, measure the absorption at 560 nm using a plate reader.
- Export the plate reader measurements and calculate the protein concentration by comparing the average absorbance values of each sample to a standard curve obtained using the protein standard. The R-squared value for the standard curve should be greater than or equal to 0.98 to accurately estimate sample protein concentration.
- Normalize the amount of protein by preparing dilutions of protein samples in sample buffer and ultrapure water. The total volume can be adjusted depending on the type of gel used. Loading 30 µg of protein per lane as a starting amount is recommended. Add reducing agent such as dithiothreitol (DTT; final concentration 5 mM) or beta-mercaptoethanol (final concentration 200 mM) to each sample as required. Pipette undiluted beta-mercaptoethanol in a fume hood.
- Incubate the samples in a heat block at 70 °C for 10 min. Put the samples on ice, vortex and spin down briefly to collect. Keep on ice until loading the gel.
- Measure the protein concentration using a bicinchoninic acid (BCA) assay. Prepare a BCA assay mix according to the manufacturer’s instructions in a 96-well optical plate using 200 µL of BCA mix per well.
2. Gel electrophoresis of protein samples
- Device and gel set up
- Setup a precast 4–12% Bis-Tris gradient gel (see Table of Materials) in the gel electrophoresis chamber system. Rinse the gels using double-distilled water before use.
NOTE: Depending on the size, interactions and abundance of the protein of interest, gels with a different gradient, buffering agent or well size and number can be used. - Add 500 mL of 1x MES SDS running buffer diluted in double-distilled water per tank. Carefully remove the comb from the gels after adding the running buffer without disturbing the wells in the stacking gel.
- Setup a precast 4–12% Bis-Tris gradient gel (see Table of Materials) in the gel electrophoresis chamber system. Rinse the gels using double-distilled water before use.
- Protein loading
- Load 3.5 µL of a protein standard into the well. Depending on the sample layout, loading a protein ladder on both sides of the gel can aid in more accurately estimating protein size. Use fine-tipped gel loading tips for more accurate sample loading.
- When using an internal standard for between-membrane normalization (see step 5 below and discussion), load an amount that is equal to the other samples into the first 3 wells next to the protein ladder.
- Load 30 µg of each sample in the remaining wells. Add 1x sample buffer to all empty wells.
- Electrophoresis
- Assemble the gel tank after loading the samples. Run the samples through the stacking gel at 80 V for 10 min followed by 150 V for an additional 45-60 min.
3. Protein transfer
NOTE: Protein transfer in this protocol is performed using a commercially available semi-dry blotting system (see Table of Materials) for fast and consistent outcomes.
- Prepare the protein transfer by pre-soaking filter paper in double-distilled water and ensuring the gel knife, plastic Pasteur pipette, blotting roller and forceps are ready to use.
- Open the transfer stack by carefully removing all wrapping foil. Remove the top from the bottom stack and set it aside. Quickly moisten the membrane on the bottom stack with several drops of electrophoresis running buffer (2-3 mL). Once the transfer stack is open, it is important to prevent the PVDF membrane from drying out.
- After stopping the electrophoresis, open the pre-cast gel using the gel knife and cut off the stacking gel. Cut the gel around its edges to free it from the plastic cast. Keep the gel knife wet to prevent damage to the gel.
- Assemble the transfer stack from bottom to top: bottom stack (containing the PVDF membrane), protein gel, filter paper. Use the blotting roller to remove all air bubbles. Place the top stack on top of the filter paper and roll the stack again to remove air bubbles. Do not push too strongly as this may cause the gel to deform during protein transfer.
- Transfer the whole stack into the transfer device with the electrode on the left side of the device and place the gel sponge on top of the stack so that it is aligned with the corresponding electrical contacts on the device. Close the lid, select and start the appropriate program (20 V for 7 min is a recommended starting point).
- When finished, leave the lid closed for 2 min to allow the stack to cool down and to prevent the membrane from drying out. Remove the transfer stack and cut the membrane to the gel size. Wash the cut membrane quickly with double-distilled water before continuing with the total protein stain.
4. Total protein staining
NOTE: Using fluorescent detection provides a substantial benefit over more traditional approaches (e.g., ECL detection), as the linear range and sensitivity can be much better controlled4. Therefore, in steps 4 and 5, a fluorescent TPS and fluorescent secondary antibodies are used (see Table of Materials).
- Roll the membrane into a 50-mL tube with the protein side facing inwards. Because of light sensitivity of the fluorescent TPS and secondary antibodies, all subsequent steps are carried out in the dark.
- Add 5 mL of protein stain solution (see Table of Materials) and incubate on a roller for 5 min at room temperature. Because TPS and wash buffer contain methanol, carry these steps out in a fume hood.
- Discard the staining solution and wash twice quickly with the 5 mL of wash solution (6.3% acetic acid in 30% methanol). Place the tube briefly back on the roller between wash steps. Rinse the membrane briefly with ultrapure water before continuing.
5. Blocking, antibody incubation and detection
- Blocking the membrane: Add 3 mL of blocking buffer (see Table of Materials) to the 50 mL tube containing the membrane. Incubate the membrane on a roller for 30 min at room temperature. Depending on the choice of antibody, the type of blocking buffer used may require optimization.
- Primary antibody incubation
- Discard the blocking buffer and replace with the primary antibody at the appropriate, optimized concentration (Figure 1 and Figure 2: mouse-anti-SMN, at 1:1,000, diluted in blocking buffer).
NOTE: Adequate optimization of primary antibodies should include confirmation that the antibody detects a protein product of the right size whilst showing no or minimal binding to other, unspecific protein products. If possible, test and compare multiple antibodies against the protein of interest. - Incubate the membrane on a roller overnight at 4 °C. The next day, remove the antibody solution and wash 6 times for 5 min with 1x PBS on a roller at room temperature (RT).
- Discard the blocking buffer and replace with the primary antibody at the appropriate, optimized concentration (Figure 1 and Figure 2: mouse-anti-SMN, at 1:1,000, diluted in blocking buffer).
- Secondary antibody incubation
- Prepare the specific secondary antibody at 1:5,000 against the host of the primary antibody in 5 mL blocking buffer. Other secondary antibodies may require other dilutions or the use of alternative blocking buffers.
- Incubate the membrane with the secondary antibody solution on a roller for 1 h at RT. After incubation, wash the membrane three times 30 min with 1x PBS on a roller.
- Dry the membrane and keep the membrane protected from light using aluminum foil until detection. Membranes can be kept at 4 °C for long term storage.
- Image acquisition
- Login to the computer attached to the scanner. Place the membrane on the scanner with the protein side facing down and select the scanning area in the software.
- Optimize the laser intensity for both (700 nm and 800 nm) channels, by confirming no saturation occurs. Acquire the images in both channels and export the images for further analysis (step 6).
6. Western blot analysis and quantification
NOTE: These recommendations are based on the freely available Image Studio software. However, comparable analyses can also be done using other software packages, such as ImageJ.
- Import the file: Create a local workspace on the computer used for analysis. This generates a database of image files for acquired western blots. Import files obtained on the scanner and select the image for analysis.
- TPS analysis
- Display the 700 nm channel to show the total protein staining result. An example of a TPS image is included in Figure 1 in which different tissues from neonatal (P5) (Figure 1A-B) and 10-week old mice (Figure 1C-D) are compared directly. Similarly, Figure 2 shows an example of a direct comparison of brain tissue from mice of different ages.
- Select Analysis tab from top right corner and select Add Rectangle to define the area of interest for normalization (Figure 1B, D). Copy and paste the first rectangle area onto each individual sample to ensure the defined region is at the same size for all analyzed lanes.
- Copy the result from the Shapes tab in the bottom left corner of the software.
- Quantification
NOTE: The optimal approach for quantifying samples depends on the experimental design. We will here provide an illustrative example of the detection of the survival motor neuron protein (Smn; a key protein involved in the neuromuscular disease spinal muscular atrophy20,21), that is known to decrease over time19 and how normalization of Smn signal intensity to TPS provides reliable estimates of protein expression development.- Figure 2 shows a decrease of Smn expression with increasing age of the animal with TPS as protein loading control. Repeat Step 6.2.1-6.2.3 to quantify protein loading (Figure 2B). Repeat Step 6.2.1-6.2.3 in the 800 nm channel (Figure 2A) to analyze the protein of interest.
- Copy the results from both TPS and protein of interest to a spreadsheet program. On the spreadsheet, first normalize the protein loading by determining the highest TPS signal and dividing each TPS signal value by this value to obtain the normalized protein loading value.
- Divide the 800 nm signal value from each individual sample by its corresponding normalized protein value to calculate the relative protein expression ratio in different samples.
- After the first normalization, compare various time points or tissues to the average value of the internal standard to allow direct comparisons across different membranes and experiments.
7. Statistics
- To determine any statistically significant differences in protein expression across complex and large groups of samples, appropriate statistical methodology is required. Although a detailed discussion of statistical background goes beyond the scope of this paper, we want to highlight several considerations and detail a successful approach that we have used previously19.
- As for many experiments, protein quantification measurements do not represent completely independent data. Here, for example, multiple tissues are generally obtained from single animals to determine protein levels across multiple organs at a single experimental time-point. Therefore, we used a mixed effects models to analyze differences in protein expression over time, and between tissues. In general, mixed effects models provide an effective means to deal with non-independence and thereby avoid pseudo-replication22,23. In the present case, mixed effects models increase statistical power by accounting for repeated measurements among tissues, within individuals. We use the statistical software package R to perform these analyses, as this is freely available and versatile. However, other commercially available packages may be able to perform similar analyses.
- The current experimental design involves a “split plot” design because each mouse belonged to only one age group. Therefore, we modeled individual mice (mouse ID) as a random effect with a unique identifier; we also accounted for tissue, age, and their interaction as fixed effects. We modeled the data using the function lmer in the R library, lme4. As a quality control step, we visualized residuals to assess the assumptions of equal variance and a normal distribution and transformed the data where necessary to meet these assumptions. To test for a significant interaction between tissue and age, we fit an identical model that lacked this interaction, and compared the models using parametric bootstrapping (R function PBmodcomp in the library, pbkrtest). Where significant interactions arose, we used the function emmeans (in the emmeans R library) to determine the cause of the interaction. For example, we compared mean expression among age classes within each tissue; a significant interaction may arise between age and tissue if, say, two given age classes differed significantly for one tissue but not another. In summary, these approaches provide a way to extend the robustness of conclusions from western blot experiments by providing statistical support to these results.
Representative Results
We include examples of the use of TPS and an internal standard to facilitate comparisons of protein levels across tissues and time points. Figure 1 shows results from Western blotting on protein extracted from tissues obtained from neonatal (postnatal day 5) in comparison to adult mice (10-week old). TPS and Smn immunoblot are shown in Figure 1A, C. Quantification of fluorescence intensity of the TPS was achieved by measuring the fluorescence intensity inside the rectangle box on each lane and its results are shown in the tables in Figure 1B and 1D. Note that samples from different tissues are characterized by different TPS protein band patterns and therefore it is necessary to use the whole lane for normalization purposes. Indeed, when whole lanes are analyzed, the fluorescence intensity remains relatively similar across samples, indicating TPS for normalization is suitable for this purpose. An internal standard consisting of a P5 brain lysate mixture was also included to illustrate how it can be used for further comparisons between different membranes. Furthermore, in Figure 2, we show how a fluorescent TPS can be used to compare protein levels at different developmental time points. Here, we show Smn levels in brain lysates from neonatal (P5), weaning age (P20) and adult (10W) mice (Figure 2A). Although Smn levels clearly decrease with age, TPS quantification remains constant as illustrated in Figure 2B.
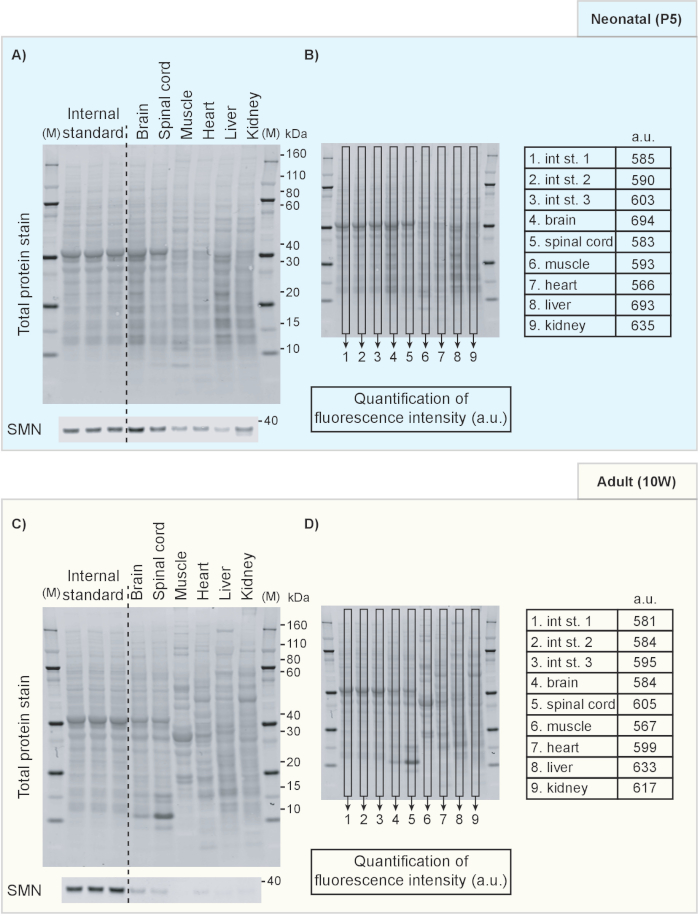
Figure 1. Western blots showing TPS and Smn protein levels in mouse tissues at two different ages. TPS and Smn protein for P5 (A) and 10 week-old (C) mice. (B, D) The fluorescence intensity of whole-lane TPS was calculated and is indicated (in arbitrary units). M: marker/protein standard; kDa: kilodalton; a.u.: arbitrary unit; P5: postnatal day 5; 10W: 10 weeks. Please click here to view a larger version of this figure.
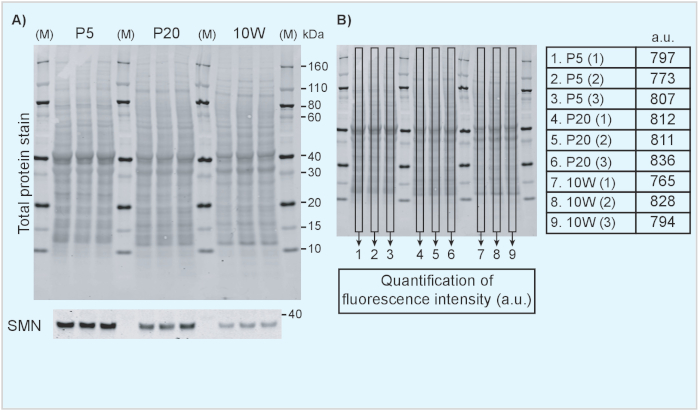
Figure 2. Analysis of Smn expression in mouse tissues at different developmental time points. (A) Brain lysates from tissue obtained from P5, P20 and 10 week-old mice was analyzed using TPS (top panel) and SMN (bottom panel). (B) The fluorescence intensity of the TPS was calculated and is indicated in arbitrary units. M: marker/protein standard; kDa: kilodalton; a.u.: arbitrary unit; P5: postnatal day 5; P20: postnatal day 20; 10W: 10 weeks. Please click here to view a larger version of this figure.
| Tissue | Approx. weight* | 1xPBS wash (4 °C) | Repeat wash | Homogenizing buffer ** |
| Spinal cord | 40 mg | 150 mL | 3x | 100 mL |
| Muscle (GC) | 20 mg | 150 mL | 3x | 100 mL |
| Brain | 240 mg | 400 mL | 4x | 400 mL |
| Heart | 60 mg | 400 mL | 4x | 200 mL |
| Liver | 130 mg | 400 mL | 4x | 400 mL |
| Kidney | 45 mg | 400 mL | 4x | 200 mL |
| * These weights are indicative values for tissue obtained from P8 mice. | ||||
| ** These volumes are indications and can be further adjusted according to the weight of the tissue. | ||||
Table 1. Overview of expected tissues weights and corresponding recommendations for PBS washes and lysis buffer volume to be used for homogenization. The weights are indications for tissue obtained from postnatal day 8 (P8) mice. PBS and lysis buffer volumes can be scaled up and down according to experimental needs.
Discussion
With the appropriate experimental design, control measures and statistical analysis, western blotting can be used to make reliable quantitative estimates of protein expression within and between a varied range of biological samples. The protocol we describe in the current manuscript aims to serve as a guideline for researchers looking to use Western blotting to undertake quantitative analysis across larger and more complex groups of samples, by using a combination of fluorescence-based detection methods, total protein loading normalization and internal standards. Although the focus here is on determining and comparing protein expression from different mouse tissues and at different ages, this approach can also be extended to compare protein expression in other experimental conditions.
A central step in our current protocol is the normalization of proteins of interest to total protein loaded by quantifying a fluorescent total protein stain. TPS normalization corrects for variation in sample loading and error margins in protein quantification methods. However, because the number of protein samples that can be analyzed on a single membrane is often limited, further normalization may be required to compare multiple membranes. Indeed, variability between how proteins are detected on different membranes (due to for example antibody incubation time or temperature variation) may cause variation beyond that introduced through loading and quantification steps of the protocol. Here, we use an internal standard that consists of a single protein lysate mix that is loaded in triplicate on each gel and allows for comparison and normalization between gels/membranes. Theoretically, this could be any protein sample, as long as it can be made in sufficient quantities so that the same sample can be used for all experiments. In our experiments, we use a mixture of brain protein lysates, as brain homogenates contain large quantities of protein and are typically obtained at a high concentration. Averaging quantification of triplicate standard should further increase the accuracy of quantifications across membranes and contribute to the reproducibility of the experiment.
Protein levels can be determined by a number of different techniques and the preferred method depends on the sample type being analyzed and the goal of the experiment. Reproducible quantification of Western blot works best in situations where experimental conditions can be well-controlled, such as when using mouse or other animal models of a defined genetic background. In contrast, in many experiments using human patient samples, this may be less feasible as age, genetic variability and tissue sampling times are much harder to control than in (animal) model systems. Plate-based techniques such as ELISA might be more suitable for these analyses, although the careful validation of antibody specificity is crucial. For example, in fragile X syndrome research, antibodies have been shown to detect different isoforms and when used in ELISA this would lead to overestimation of the total amount of protein as ELISA determines a signal for all isoforms combined24. Optimal choices for methods to determine protein expression are therefore depending on context, sample type and the research question that is being investigated.
Performing adequate statistical analysis is a prerequisite for the reliability of any conclusions drawn from the quantification of biological data. Statistically analyzing complex data as generated by comparing different tissues, time points or other experimental conditions and combinations thereof may require more advanced statistical modelling than ANOVA with post-hoc testing can deliver. For more complex statistical modelling, such as the mixed-effects model approach, we describe in the current manuscript, it may be advisable to seek further advise from biostatisticians. Adequate statistical analysis of large-scale protein expression can greatly improve the robustness of the outcomes and the reliability and reproducibility of results.
In summary, the experimental approach we describe here provides a robust and reproducible method for researchers that want to determine protein expression using western blot in complex samples allowing to answer new and exciting research questions.
Divulgations
The authors have nothing to disclose.
Acknowledgements
E.J.N.G. is supported by the Wellcome Trust (grant 106098/Z/14/Z). Other funding has been provided by the SMA Trust (SMA UK Consortium; T.H.G. & Y-T.H.), SMA Europe (T.H.G, D.v.D.H. & E.J.N.G.), the University of Edinburgh DTP in Precision Medicine (T.H.G., L.L. & A.M.M.), and the Euan MacDonald Centre for Motor Neurone Disease Research (T.H.G).
Materials
| Fine Tipped Gel Loading Tips | Alpha Laboratories | GL20057SNTL | |
| Halt Protease Inhibitor Cocktail, EDTA-free 100x 5mL | ThermoFisher Scientific | 78437 | |
| Handheld homogeniser | VWR Collection | 431-0100 | |
| iBlot 2 Gel Transfer Device | ThermoFisher Scientific | IB21001 | |
| iBlot Transfer Stack, PVDF, regular size | ThermoFisher Scientific | IB401031 | |
| Image Studio Lite | Licor | N/A | Free download from https://www.licor.com/bio/products/software/image_studio_lite/ |
| IRDye 800CW secondary antibodies | Licor | — | Select appropriate secondary antibody that is specific against host of primary antibody. |
| Micro BCA Protein Assay Kit | ThermoFisher Scientific | 23235 | |
| Novex Sharp Pre-stained Protein Standard | ThermoFisher Scientific | LC5800 | |
| NuPAGE 4-12% Bis-Tris Protein Gels, 1.0 mm, 15-well | ThermoFisher Scientific | NP0323BOX | |
| NuPAGE LDS Sample Buffer (4X) | ThermoFisher Scientific | NP0007 | |
| NuPAGE MOPS SDS Running Buffer (20X) | ThermoFisher Scientific | NP0001 | |
| Odyssey Blocking Buffer | Licor | 927-40000 | |
| Purified Mouse anti-SMN (survival motor neuron) monoclonal antibody | BD Transduction Laboratories | 610646 | Is used extensively in the SMN/SMA literature and gives consistent results regardsless of lot number |
| REVERT Total Protein Stain, 250 mL | Licor | 926-11021 | |
| REVERT Wash Solution | Licor | 926-11012 | |
| RIPA Lysis and Extraction Buffer | ThermoFisher Scientific | 89900 | |
| XCell SureLock Mini-Cell | ThermoFisher Scientific | EI0001 |
References
- Bertoni, T. A., Perenha-Viana, M. C., Patussi, E. V., Cardoso, R. F., Svidzinski, T. I. Western blotting is an efficient tool for differential diagnosis of paracoccidioidomycosis and pulmonary tuberculosis. Clinical and Vaccine Immunology. 19 (11), 1887-1888 (2012).
- Hutchinson, A. B., et al. Costs and outcomes of laboratory diagnostic algorithms for the detection of HIV. Journal of Clinical Virology. 58 Suppl 1, (2013).
- Towbin, H., Staehelin, T., Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America. 76 (9), 4350-4354 (1979).
- Eaton, S. L., et al. Total protein analysis as a reliable loading control for quantitative fluorescent Western blotting. PloS One. 8 (8), e72457 (2013).
- Ghosh, R., Gilda, J. E., Gomes, A. V. The necessity of and strategies for improving confidence in the accuracy of western blots. Expert Rev Proteomics. 11 (5), 549-560 (2014).
- Jensen, B. C., Swigart, P. M., Simpson, P. C. Ten commercial antibodies for alpha-1-adrenergic receptor subtypes are nonspecific. Naunyn-Schmiedebergs Archives of Pharmacology. 379 (4), 409-412 (2009).
- Jositsch, G., et al. Suitability of muscarinic acetylcholine receptor antibodies for immunohistochemistry evaluated on tissue sections of receptor gene-deficient mice. Naunyn-Schmiedebergs Archives of Pharmacology. 379 (4), 389-395 (2009).
- Smejkal, G., Gallagher, S. Determination of semidry protein transfer efficiency with transverse gradient gel electrophoresis. Biotechniques. 16 (2), (1994).
- Eaton, S. L., et al. A guide to modern quantitative fluorescent western blotting with troubleshooting strategies. Journal of Visualized Experiments. (93), e52099 (2014).
- Hunter, G., Roche, S. L., Somers, E., Fuller, H. R., Gillingwater, T. H. The influence of storage parameters on measurement of survival motor neuron (SMN) protein levels: implications for pre-clinical studies and clinical trials for spinal muscular atrophy. Neuromuscular Disorders. 24 (11), 973-977 (2014).
- Fosang, A. J., Colbran, R. J. Transparency Is the Key to Quality. Journal of Biological Chemistry. 290 (50), 29692-29694 (2015).
- Taylor, S. C., Berkelman, T., Yadav, G., Hammond, M. A defined methodology for reliable quantification of Western blot data. Molecular Biotechnology. 55 (3), 217-226 (2013).
- Goasdoue, K., Awabdy, D., Bjorkman, S. T., Miller, S. Standard loading controls are not reliable for Western blot quantification across brain development or in pathological conditions. Electrophoresis. 37 (4), 630-634 (2016).
- Rocha-Martins, M., Njaine, B., Silveira, M. S. Avoiding pitfalls of internal controls: validation of reference genes for analysis by qRT-PCR and Western blot throughout rat retinal development. PloS One. 7 (8), e43028 (2012).
- Aghamaleky Sarvestany, A., et al. Label-free quantitative proteomic profiling identifies disruption of ubiquitin homeostasis as a key driver of Schwann cell defects in spinal muscular atrophy. Journal of Proteome Research. 13 (11), 4546-4557 (2014).
- Fuller, H. R., et al. Spinal Muscular Atrophy Patient iPSC-Derived Motor Neurons Have Reduced Expression of Proteins Important in Neuronal Development. Frontiers in Cellular Neuroscience. 9, 506 (2015).
- Liu, N. K., Xu, X. M. beta-tubulin is a more suitable internal control than beta-actin in western blot analysis of spinal cord tissues after traumatic injury. Journal of Neurotrauma. 23 (12), 1794-1801 (2006).
- Moritz, C. P. Tubulin or Not Tubulin: Heading Toward Total Protein Staining as Loading Control in Western Blots. Proteomics. 17 (20), (2017).
- Groen, E. J. N., et al. Temporal and tissue-specific variability of SMN protein levels in mouse models of spinal muscular atrophy. Human Molecular Genetics. 27 (16), 2851-2862 (2018).
- Chaytow, H., Huang, Y. T., Gillingwater, T. H., Faller, K. M. E. The role of survival motor neuron protein (SMN) in protein homeostasis. Cellular and Molecular Life Sciences. , (2018).
- Groen, E. J. N., Talbot, K., Gillingwater, T. H. Advances in therapy for spinal muscular atrophy: promises and challenges. Nature Reviews: Neurology. 14 (4), 214-224 (2018).
- Fitzmaurice, G. M., Laird, N. M., Ware, J. H. . Applied longitudinal analysis. , (2004).
- Jordan, C. Y. Population sampling affects pseudoreplication. PLoS Biology. 16 (10), e2007054 (2018).
- LaFauci, G., Adayev, T., Kascsak, R., Brown, W. T. Detection and Quantification of the Fragile X Mental Retardation Protein 1 (FMRP). Genes (Basel). 7 (12), (2016).
