The following observations demonstrate successful selection-amplification (SELEX). First, we analyzed pool 0 and the selected sequences for binding to the protein used for the iterative selection-amplification approach. Figure 2 shows that the mammalian polypyrimidine-tract binding protein (PTB) shows barely detectable binding to the pool 0 sequence but high affinity for the selected sequence pool. There was barely detectable binding to pool 0 when we used about 300-fold higher protein concentration for binding than used for the selected pool. Thus, there was at least a several hundred-fold difference in protein binding affinities between the random or starting pool and the selected pool. This observation experimentally confirms that the selection-amplification protocol described here is successful.
Second, we sequenced the selected pool and determined a consensus binding site. The consensus sequence obtained from alignment of the majority of selected sequences from the mammalian PTB-selected pool was: GCCUG(Y/G)UGCYYYYCYYYG(Y/G)CCC. This shows that we have selected unique pyrimidine-rich sequences that bind PTB11. When we performed iterative selection-amplification for the RNA-binding domain of the Drosophila PTB, we enriched CU-rich sequences interrupted by guanosines. Among the high affinity sequences that the Drosophila PTB selected was an 84% pyrimidine-rich sequence: GCUUUCCUCUGUCGCCCUUCUUCGUCCCCUG. In fact, this sequence is similar to the pyrimidine-rich sequence present in the alpha-tropomyosin intron which binds with high affinity to and is regulated by the mammalian PTB15. We have successfully used this approach repeatedly to study RNA-binding properties and functions splicing regulators and a splicing factor11, 15, 16. Table 1 shows successful examples of RNA-binding proteins for which SELEX was used to identify their preferred or consensus binding site(s).
Third, an in vitro splicing assay, which is based on alternative 3' splice site choice, shows functional relevance of distinct but overlapping RNA-binding specificities of polypyrimidine-tract binding proteins. Whereas an upstream 3' splice site is used by default, addition of the recombinant PTB leads to activation of the alternative or downstream 3' splice site (Figure 3). In contrast, addition of recombinant hnRNP C17 leads to repression of both 3' splice sites. Addition of the recombinant general splicing factor U2AF65 reverses the hnRNP C1-mediated 3' splice site repression (Figure 3) as well as the PTB-mediated effect on downstream 3' splice site activation (data not shown). A simple explanation for these effects is a direct competition between the binding of the general splice factor U2AF65 and PTB (also called hnRNP I), which preferentially binds to and represses certain 3' splices sites, or between U2AF65 and hnRNP C, which binds to and represses both 3' splice sites.
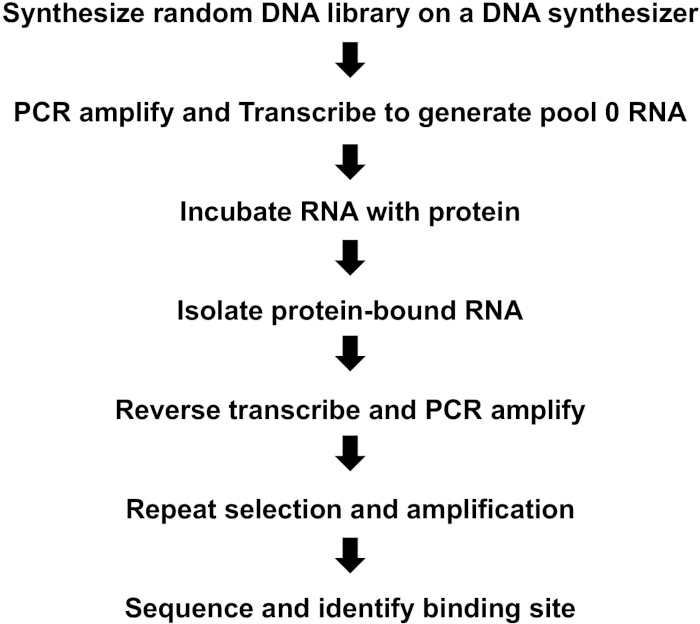
Figure 1: Summary of key steps in iterative selection-amplification process (SELEX). Please click here to view a larger version of this figure.
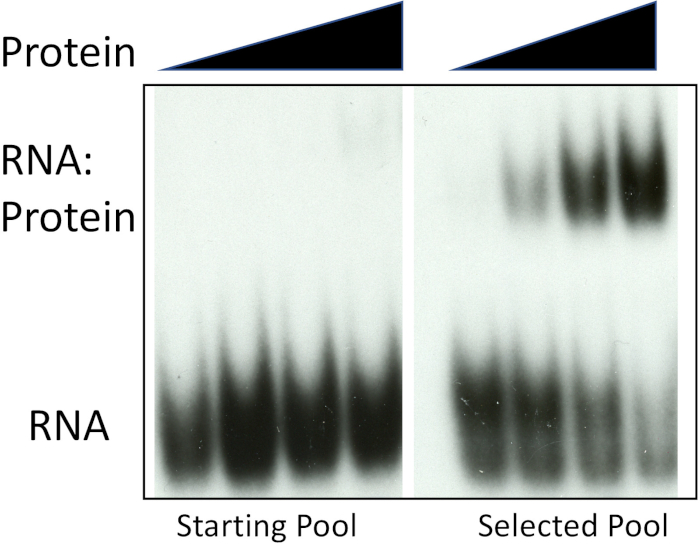
Figure 2: Enrichment of PTB-binding RNAs. Increasing concentration (filled triangles) of recombinant PTB was used with either radiolabeled pool 0 RNA or the selected pool obtained following six rounds of selection and amplification. Positions of unbound RNA and the RNA:protein complex are indicated. Please click here to view a larger version of this figure.
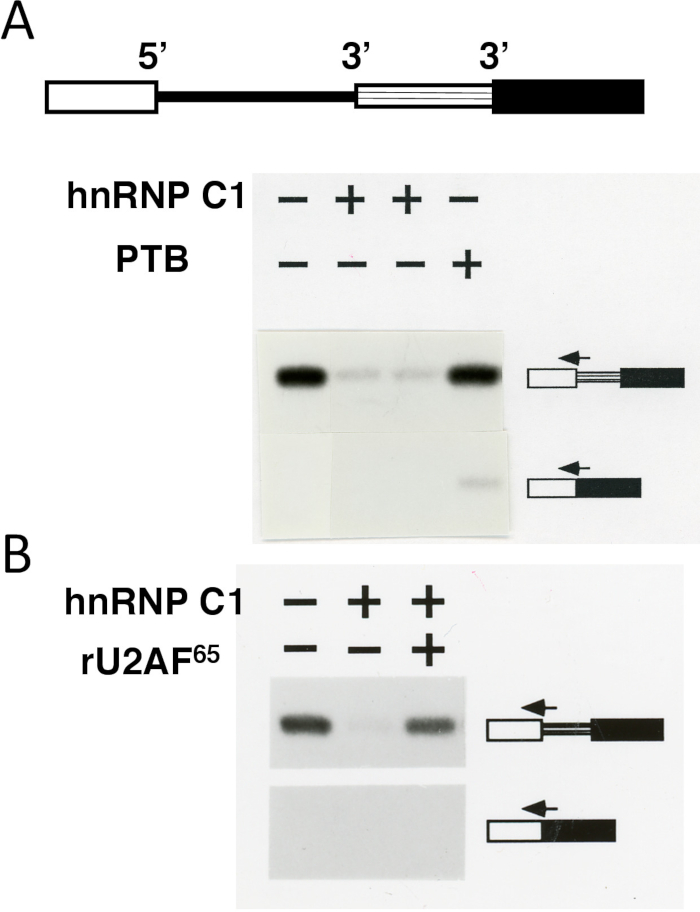
Figure 3: Splice site switching assay validates distinct binding specificities of pyrimidine-binding proteins. (A-Top) Schematics of the splicing substrate. The splicing substrate contains a 5' splice site and two alternative 3' splice sites flanking the intron. Rectangles (open, with horizontal lines, and solid) are exons and the line is an intron. (A-Bottom) hnRNP C1 represses the upstream 3' splice site (without activation of the downstream 3' splice site), whereas PTB leads to activation of the downstream 3' splice site. The splicing substrate was incubated in a HeLa cell nuclear extract.The splicing products (shown on the sides) were analyzed using a primer extension assay18 with splice-junction primers (arrows), which recognize splicing of the common 5' splice site to either the upstream or the downstream 3' splice site. (B) Recombinant U2AF65 (rU2AF65) reverses the repressive effect of hnRNP C1. Addition of the recombinant hnRNP C1, PTB, or rU2AF65 proteins to the splicing reaction is indicated by the + symbols. Please click here to view a larger version of this figure.
| Protein | Preferred sequence(s) |
| U2AF65 | U-rich containing Cs |
| SXL | U-rich containing 2-4 Gs |
| PTB | UCUUC-rich with some Gs |
| hnRNP C1 | U-rich (5-6 long) |
| CstF64 | GU-rich |
| hnRNP E1/E2 and K | C-rich |
| U2AF65/U2AF35 heterodimer | UUUYYYYUNUAGGU |
Table 1: Preferred binding sites for some RNA-binding proteins.
