P. andersonii trees can be grown in a conditioned greenhouse at 28 °C and ~85% relative humidity (Figure 1A). Under these conditions, trees start flowering at 6-9 months after planting. Female P. andersonii flowers produce berries that each contains a single seed. During maturation, the berries change color; first from green to white and subsequently from white to brown (Figure 1B). Seeds extracted from the ripened brown berries, germinate well after a 10-day temperature cycle and a 7-day incubation on SH-0 plates (Figure 1C). Germinated seeds continue to develop into young seedlings that can be used for experimentation after ~4 weeks (Figure 1D).
We have previously shown that petioles and segments of young P. andersonii stems can be efficiently transformed using A. tumefaciens strain AGL110. At the start of the transformation procedure, the tissue explants are co-cultivated with A. tumefaciens for 2 days at 21 °C (Figure 2A). Prolonged co-cultivation results in the over-colonization of the tissue explants by A. tumefaciens and should, therefore, be prevented (Figure 2B). After the co-cultivation period, tissue explants are transferred to selective media, which promotes outgrowth of transformed tissue. Two to three weeks later, small green micro-calli are generally observed along the original wound surface (Figure 2C). These calli should continue to grow and develop 1 or more putatively-transformed shoots at 6-8 weeks after the transformation procedure has been initiated (Figure 2D). At this stage, transformation efficiencies typically range from ~10-30% for transformations initiated with tissue explants taken from mature and partly woody branches (Table 7). If transformations are initiated with explants taken from the young and rapidly-growing tips of branches that are not yet bearing flowers, transformation efficiencies of ~65-75% can be achieved (Table 7). Occasionally, whitish calli are formed on the side of an explant that is not in contact with the medium and, therefore, do not experience kanamycin selection. These calli are often not transgenic and any shoots formed from these calli will generally bleach and die after direct contact with kanamycin-containing medium (Figure 2E). In case the transformation rate is low and/or the starting material was suboptimal, tissue pieces might turn brown (Figure 2F) and suffer from over-proliferation by A. tumefaciens (Figure 2G). To prevent A. tumefaciens from spreading and overgrowing nearby explants, regular refreshment of the medium is required, and severely infected explants need to be removed. Once individual transgenic shoots are placed in the propagation medium, over-proliferation by A. tumefaciens is generally not occurring anymore (Figure 2H). Transgenic shoots can be multiplied through in vitro propagation, which will give rise to tens of shoots in a period of one month (Figure 3A-B). These shoots can be placed on rooting medium, which should induce root formation after ~2 weeks (Figure 3C-D). Rooted plantlets can be subsequently used for experimentation.
To create knockout mutant lines, we make use of CRISPR/Cas9-mediated mutagenesis. To this end, we make use of a binary vector containing the kanamycin resistance gene NPTII, a Cas9-encoding sequence driven by the CaMV35S promoter and 2 sgRNAs per target gene that are expressed from the AtU6p small RNA promoter20. A graphical representation of the construct used for CRISPR/Cas9-mediated mutagenesis of P. andersonii is provided in Figure 4A. Using this method, genome editing is observed in ~40% of putatively-transformed shoots10. To identify mutant lines, putatively-transformed shoots are genotyped for mutations at the sgRNA target site(s) using primers spanning the targeted region. An example of the expected results is given in Figure 4. As can be seen from the photo taken after gel electrophoresis, several samples produce a PCR amplicon with similar size to the wild type (Figure 4B). These plants may contain small indels that cannot be visualized by agarose gel electrophoresis or remain unedited by the Cas9 enzyme. Additionally, several samples yield bands that are different in size from the wild type (e.g., lines 2, 4, 7 and 8 in Figure 4B). In these lines, 1 (lines 4, 7 and 8) or both (line 2) alleles contain larger indels that can be easily visualized. The exact nature of the mutations at the target site(s) is revealed after PCR amplicon sequencing. As can be seen from Figure 4C, both small indels of 1-4 bp, as well as, larger deletions can be obtained after CRISPR/Cas9 mutagenesis. In Figure 4C, the sequence of line 1 is identical to that of the wild type, indicating that this line escaped editing and, therefore, should be discarded. Among the lines that contain mutations, heterozygous, homozygous and bi-allelic mutants can be identified (Figure 4C). However, heterozygous mutants are generally rare10. Homozygous or bi-allelic knockout mutants can be propagated vegetatively to obtain sufficient material for phenotypic analysis. As phenotypic analysis is performed in the T0 generation, it is important to check whether mutant lines might be chimeric. To this end, genotyping needs to be repeated on at least 3 different samples taken from each mutant line. If the genotyping results are identical to each other and the original genotyping sample (e.g., line 8 in Figure 4D), the line is homogeneously mutated and can be used for further analysis. However, if the genotyping results differ between independent samples (e.g., line 4 in Figure 4D), the mutant line is chimeric and needs to be discarded.
Inoculation of P. andersonii with M. plurifarium BOR2 results in the formation of root nodules (Figure 5). As can be seen in Figure 5A, these nodules are distributed along the root system. Nodules of P. andersonii are light brown in color but can be easily discriminated from the root tissue based on their shape (Figure 5B). Inoculation experiments in pots and subsequent growth for 4-6 weeks typically result in the formation of ~10-30 nodules (Figure 6A). A similar number of nodules is formed after inoculation of EKM plate-grown P. andersonii plantlets at 4 weeks after inoculation (Figure 6A). In pouches, P. andersonii seedlings typically form ~5-15 nodules at 5 weeks post inoculation (Figure 5C-D, 6A). To analyze the nodule cytoarchitecture, nodules can be sectioned and observed using bright-field microscopy. Figure 6B shows an example of a longitudinal section through the middle of a P. andersonii nodule. This section shows the central vascular bundle of a P. andersonii nodule, which is flanked by nodule lobes containing infected cells (Figure 6B).
P. andersonii plantlets can also be mycorrhized. After 6 weeks of inoculation with R. irregularis, mycorrhizal colonization frequency typically reaches > 80% (Figure 6C). At this time point, generally ~30% of the cells contain arbuscules (Figure 6C). A representative image of a P. andersonii root segment containing arbuscles is shown in Figure 6D.

Figure 1: Representative images of a P. andersonii tree, seeds and seedlings. (A) Six-month old P. andersonii tree grown in potting soil in a greenhouse conditioned at 28 °C. (B) Representative image depicting P. andersonii berries at various stages of maturation. Young P. andersonii berries (unripe) will change color from green to white and finally to brown (ripe) upon ripening. (C) P. andersonii seeds incubated on SH-0 medium for 1 week. A black circle indicates a germinated seedling. (D) Four-week old P. andersonii seedlings grown in SH-0 medium. Scale bars are equal to 25 cm in (A) and 1 cm in (B-D). Please click here to view a larger version of this figure.
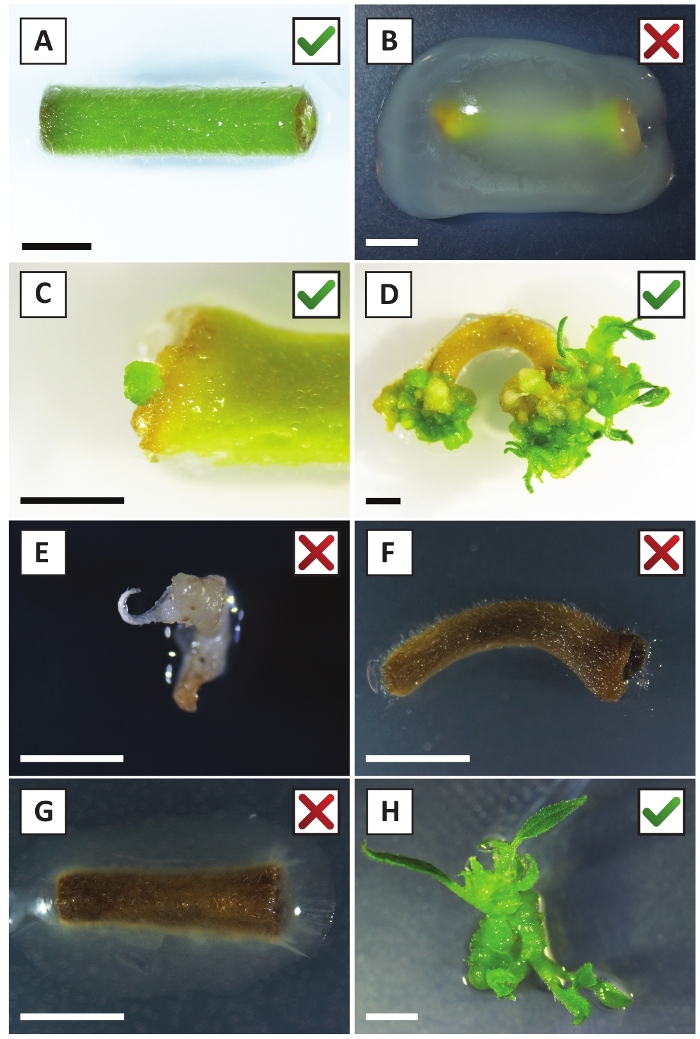
Figure 2: Representative images of explants at different stages of the stable transformation procedure. (A) Explant co-cultivated with A. tumefaciens. (B) Explant overgrown by A. tumefaciens during the first 2 weeks post transformation. (C) Transgenic micro-callus formed near the wound site of an explant at 2.5 weeks post co-cultivation. (D) Representative image of an explant at 6 weeks post co-cultivation showing the emergence of shoots from (transgenic) calli. (E) Representative image of a shoot that becomes whitish and eventually dies when in direct contact with kanamycin-containing medium. This shoot is most likely non-transgenic and escaped kanamycin selection when attached to the explant. (F) Representative image of an unsuccessfully transformed explant. (G) Representative image of an unsuccessfully transformed explant overgrown by A. tumefaciens. (H) Single transgenic shoot grown on propagation medium at 8 weeks post co-cultivation with A. tumefaciens. Scale bars equal 2.5 mm. Boxes containing green check marks or red crosses indicate successful or unsuccessful transformation of explants, respectively. Please click here to view a larger version of this figure.

Figure 3: Representative images of in vitro propagation. (A) Shoots grown on propagation medium. The image was taken 1 week after plates were refreshed. (B) Shoots grown on propagation medium. The image was taken 4 weeks after plates were refreshed. (C) Freshly cut shoots placed on rooting medium. (D) Shoots incubated on rooting medium for 2 weeks. Note the presence of roots. Scale bars are equal to 2.5 cm. Please click here to view a larger version of this figure.
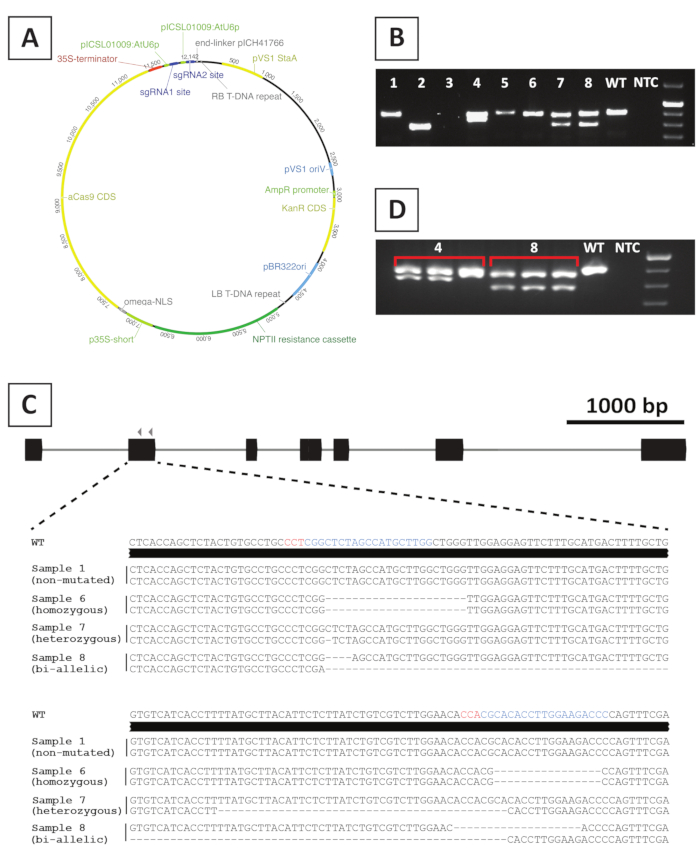
Figure 4: Representative results after genotyping of P. andersonii T0 transgenic CRISPR/Cas9 mutant lines. (A) Representative map of a binary vector used for CRISPR/Cas9-mediated mutagenesis of P. andersonii. (B) Representative result after PCR-based genotyping of potential CRISPR/Cas9 mutant lines using primers spanning the sgRNA target site(s). Shown is an image after agarose gel electrophoresis of amplicons. Samples taken from individual transgenic lines are indicated by numbers. Wild type (WT) and no template control (NTC) indicate lanes containing positive and negative controls, respectively. (C) Schematic representation of mutant alleles obtained after CRISPR/Cas9-mediated gene editing. Highlighted in blue and red colors are the sgRNA target sites and PAM sequences, respectively. (D) Representative result after PCR-based screening for potential chimeric mutant lines. Shown is an image after agarose gel electrophoresis of 3 individual samples taken from mutant lines 4 and 8. Note that transgenic mutant line 4 is chimeric. Please click here to view a larger version of this figure.
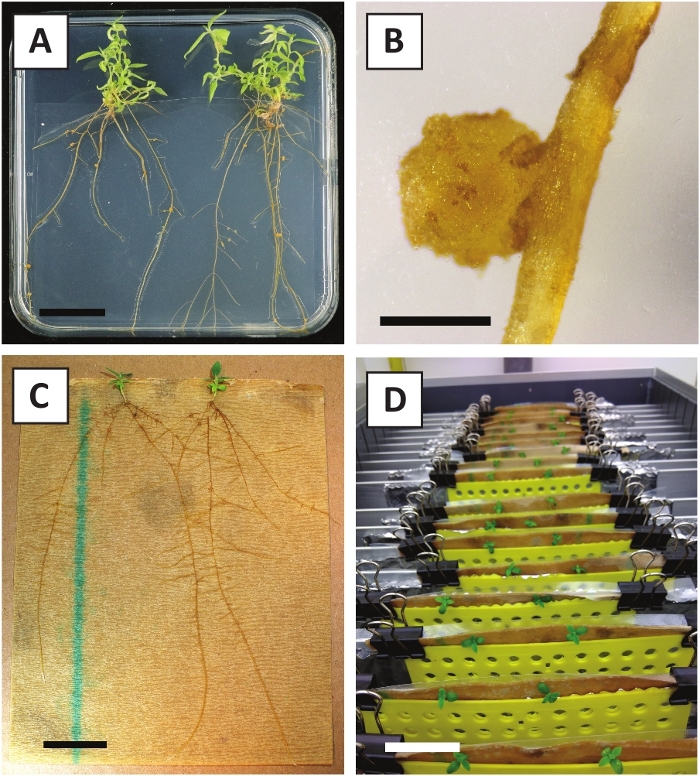
Figure 5: Representative images of nodulation assays in plates and pouches. (A) Nodulation on plates containing agar-solidified EKM medium and inoculated with M. plurifarium BOR2 for 4 weeks. (B) Representative image of a P. andersonii root nodule. The image was taken at 4 weeks post inoculation with M. plurifarium BOR2. (C) Nodulation in pouches containing liquid EKM medium. Seedlings were inoculated with Bradyrhizobium sp. Kelud2A4 for 5 weeks. (D) Representative image of a complete setup used for the nodulation in pouches. Scale bars are equal to 2.5 cm in (A,C), 1 mm in (B), and 5 cm in (D). Please click here to view a larger version of this figure.
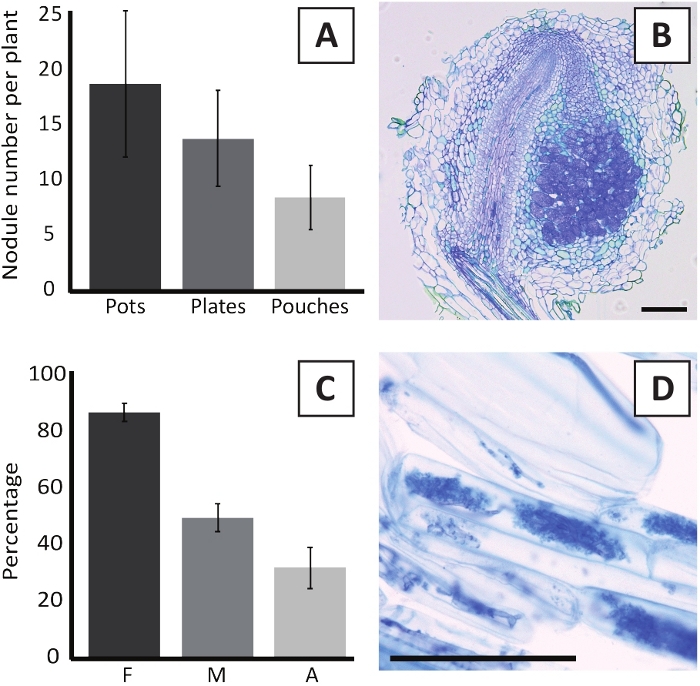
Figure 6: Representative results of the nodulation and mycorrhization assays. (A) Representative bar graph showing the number of nodules formed per plant at 4 weeks post inoculation with M. plurifarium BOR2 in pots or on plates and at 5 weeks post inoculation with Bradyrhizobium sp. Kelud2A4 in pouches. Data represent mean ± SD (n = 10). (B) Representative image of a longitudinal section through a nodule formed at 4 weeks post inoculation with M. plurifarium BOR2. The section is stained with toluidine blue. (C) Representative bar graph showing quantification of mycorrhization. Variables quantified according to Trouvelot et al.29 are F, the frequency of analyzed root fragments that are mycorrhized; M, the intensity of infection; A, the abundance of mature arbuscules in the total root system. Mycorrhization was quantified at 6 weeks post inoculation with R. irregularis (strain DAOM197198). Data represent mean ± SD (n = 10). (D) Representative image of mature arbuscules present in P. andersonii root cortical cells at 6 weeks post inoculation with R. irregularis. Scale bars equal 75 µm. Please click here to view a larger version of this figure.
| Compound | SH-0 | SH-10 | Propagation medium | Rooting medium | Infiltration medium |
| SH-basal salt medium | 3.2 g | 3.2 g | 3.2 g | 3.2 g | 3.2 g |
| SH-vitamin mixture | 1 g | 1 g | 1 g | 1 g | 1 g |
| Sucrose | – | 10 g | 20 g | 10 g | 10 g |
| BAP (1 mg/mL) | – | – | 1 mL (4.44 µM) | – | – |
| IBA (1 mg/mL) | – | – | 100 µL (0.49 µM) | 1 mL (4.92 µM) | – |
| NAA (1 mg/mL) | – | – | – | 100 µL (0.54 µM) | – |
| 1 M MES pH=5.8 | 3 mL | 3 mL | 3 mL | 3 mL | 3 mL |
| 1 M KOH | Adjust pH to 5.8 | Adjust pH to 5.8 | Adjust pH to 5.8 | Adjust pH to 5.8 | Adjust pH to 5.8 |
| Daishin agar | 8 g | – | 8 g | 8 g | – |
Table 1: Composition of Schenk-Hildebrandt-based30 media used for growing P. andersonii seedlings, stable transformation, and in vitro propagation. Dissolve solid compounds into 750 mL of ultra-pure water before adding liquid stocks. Afterwards, fill the complete medium to 1 L. Prepare BAP, IBA, NAA stocks in 0.1 M KOH and store at -20 ᵒC.
| Before autoclaving: | ||
| Compound | Amount per liter | Final concentration |
| Mannitol | 5 g | 27.45 mM |
| Na-Gluconate | 5 g | 22.92 mM |
| Yeast extract | 0.5 g | – |
| MgSO4·7H2O | 0.2 g | 0.81 mM |
| NaCl | 0.1 g | 1.71 mM |
| K2HPO4 | 0.5 g | 2.87 mM |
| After autoclaving: | ||
| Compound | Amount per liter | Final concentration |
| 1.5 M CaCl2 | 1 mL | 1.5 mM |
Table 2: Composition of Yeast-Mannitol (YEM) medium used for growing rhizobium. Adjust the pH to 7.0 and fill with ultra-pure water to 1 L. To prepare the agar-solidified YEM medium, add 15 g of microagar before autoclaving.
| Before autoclaving: | |||
| Compound | Stock concentration | Amount per liter medium | Final concentration |
| KH2PO4 | 0.44 M | Add 2 mL | 0.88 mM |
| K2HPO4 | 1.03 M | Add 2 mL | 2.07 mM |
| 500x micro-elements stock solution | – | Add 2 mL | – |
| MES pH=6.6 | 1 M | Add 3 mL | 3 mM |
| HCl | 1 M | Adjust pH to 6.6 | – |
| Ultra-pure water | – | Fill to 990 mL | – |
| After autoclaving: | |||
| Compound | Stock concentration | Amount per liter medium | Final concentration |
| MgSO4·7H2O | 1.04 M | 2 mL | 2.08 mM |
| Na2SO4 | 0.35 M | 2 mL | 0.70 mM |
| NH4NO3 | 0.18 M | 2 mL | 0.36 mM |
| CaCl2·2H2O | 0.75 M | 2 mL | 1.5 mM |
| Fe(III)-citrate | 27 mM | 2 mL | 54 μM |
Table 3: Composition of 1 L modified EKM medium31 used for P. andersonii nodulation assay. The composition of the 500x micro-elements stock solution is listed in Table 4. To prepare 2% agar-solidified EKM medium, add 20 g of Daishin agar before autoclaving. Autoclave the MgSO4·7H2O, Na2SO4, CaCl2·2H2O, and Fe(III)-citrate stocks to sterilize. Filter sterilize NH4NO3 stock solution to sterilize.
| Compound | Amount per liter | Stock concentration |
| MnSO4 | 500 mg | 3.31 mM |
| ZnSO4·7H2O | 125 mg | 0.43 mM |
| CuSO4·5H2O | 125 mg | 0.83 mM |
| H3BO3 | 125 mg | 2.02 mM |
| Na2MoO4·2H2O | 50 mg | 0.21 mM |
Table 4: Composition of the 500x micro-elements stock solution used for preparing modified EKM medium. Store the micro-elements stock solution at 4 °C.
| Compounds | Stock concentration | Amount per liter medium | Final concentration |
| K2HPO4 | 20 mM | 1 mL | 0.2 mM |
| NH4NO3 | 0.28 M | 10 mL | 2.8 mM |
| MgSO4 | 40 mM | 10 mL | 0.4 mM |
| K2SO4 | 40 mM | 10 mL | 0.4 mM |
| Fe(II)-EDTA | 9 mM | 10 mL | 0.9 mM |
| CaCl2 | 80 mM | 10 mL | 0.8 mM |
| 50x micro-elements stock solution | – | 10 mL | – |
Table 5: Composition of ½-Hoagland32 medium used for mycorrhization assays. The composition of the 50x micro-elements stock solution is listed in Table 6. Prepare the Fe(II)-EDTA solution by combining FeSO4·7H2O (9 mM) and Na2·EDTA (9 mM) into 1 stock solution, and store at 4 °C. Adjust the pH of the medium to 6.1 using 1 M KOH and fill with ultra-pure water to 1 L.
| Compounds | Amount per liter | Stock concentration |
| H3BO3 | 71.1 mg | 1.15 mM |
| MnCl2·4H2O | 44.5 mg | 0.22 mM |
| CuSO4·5H2O | 3.7 mg | 23.18 µM |
| ZnCl2 | 10.2 mg | 74.84 µM |
| Na2MoO4·2H2O | 1.2 mg | 4.96 µM |
Table 6: Composition of the 50x micro-elements stock solution used for preparing ½-Hoagland medium.
| Age of explants | Transformation efficiency |
| Young | 69.4 ± 6.2% (n = 2) |
| Mature | 18.3 ± 10.2% (n = 15) |
Table 7: Transformation efficiency of P. andersonii. Here, transformation efficiency is defined as the percentage of explants that form at least 1 transgenic callus or shoot. Transformation efficiency was scored at 6 weeks post transformation and is depicted as mean ± SD. n indicates the number of transformation experiments from which the transformation efficiency was determined.
Supplemental File 1: Overview of level 1 and level 2 constructs used for CRISPR/Cas9 mutagenesis. Please click here to download this file.
