Using the AAV system to transform normal Cas9 cells
Figure 1 provides a detailed vector map of the AAV transgene plasmid used in this study. Figure 2 outlines the design and working of the AAV-Cas9 based-system. To produce viral particles, the HEK293T cells were transfected with the AAV transgene vector and other viral packaging vectors using the PEI transfection method. After transfection, the virus-containing cells were collected and lysed, and using the heparin-agarose purification process, the viral particles were purified and concentrated (Figure 2A). These purified viral particles were used for the in vitro transformation of primary cells isolated from Cas9 mice to check the efficacy. In our system, the AAV transgene expression cassette is introduced through homologous recombination into the Cre-dependent Cas9 Rosa26 targeting template within mouse cells that carry an ubiquitous CAG promoter (pCAG), a LoxP-Stop-LoxP cassette (LSL), a Cas9 nuclease gene, a self-cleaving P2A peptide, and the reporter gene (EGFP) flanked by two homologous arms14. The Cre recombinase activity of the transduced cells excises the LSL and induces Cas9 and GFP expression (Figure 2B). Cas9 nuclease activity introduces a double-stranded break at the target sites of interest, directed through gRNAs, and helps in gene editing. The AAV system used in this study helps in delivering multiplexed gRNAs (sgRNAs for KRAS, p53, and APC) and also in expressing an oncogenic KRASG12D allele through homology-directed repair (HDR) in the transduced cells.
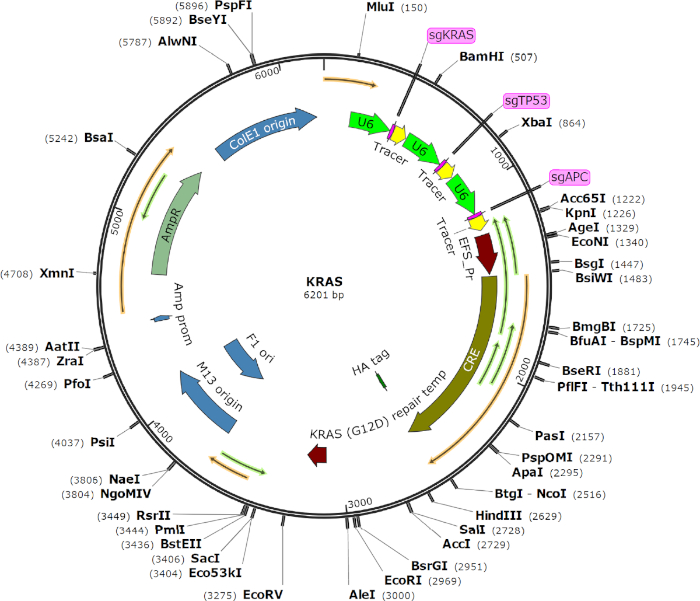
Figure 1: Vector map of the AAV transgene plasmid used in this study. This AAV system has an AAV backbone that expresses Cre recombinase and three U6-driven sgRNAs targeting KRAS, p53, and APC. It also contains a KRAS G12D homology-directed repair (HDR) donor. Please click here to view a larger version of this figure.
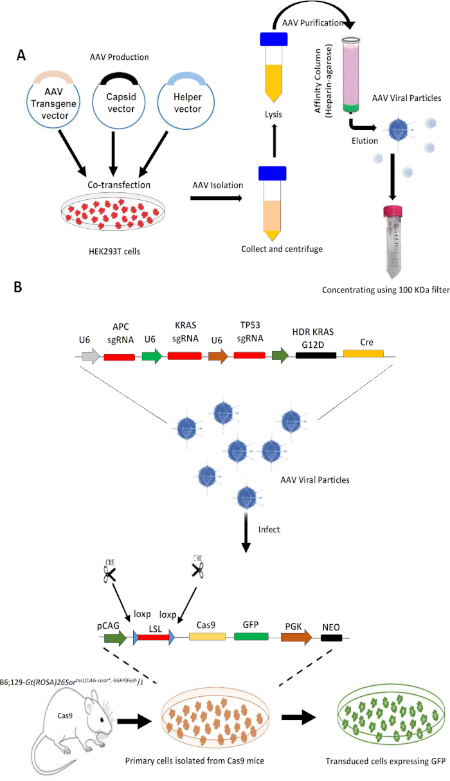
Figure 2: Design and working of a AAV-Cas9 system in transforming normal cells. (A) Outline of the production and purification of AAV particles from HEK293T after transfecting them with the AAV transgene and packaging plasmids. (B) Viral transduction of primary cells with AAV particles and the mechanism by which Cre-dependent Cas9 expression enables CRISPR editing and the transformation of primary cells to tumorigenic cells. A single AAV vector integrating gRNAs for three different genes with a knockin of the KRAS G12D allele through HDR with a Cre recombinase expression cassette was delivered into Cre-dependent Cas9 mouse primary cells. Cre recombinase activity in the transduced cells leads to excision of the LoxP-Stop-LoxP cassette and induces Cas9 and GFP expression. Please click here to view a larger version of this figure.
Validation of the AAV-Cas9 system in vitro in isolated mouse embryonic fibroblast cells
Primary mouse embryonic fibroblast (MEF) cells have been used exclusively to study the consequences of gene ablation in cellular growth and proliferation22. To validate the efficiency of the AAV-Cas9 system in transforming normal cells, we transduced MEF cells isolated from Cas9 mouse embryos (E14) with AAV. The MEF cells were isolated as described previously23,24. MEFs from 14-day-old Cas9 mouse embryos were used because they are easy to culture and transduce in vitro. In brief, to transform MEFs via AAV transduction, primary MEFs at approximately 30% confluence were considered ready for transduction. One hour before transduction, the MEF culture media was changed. The purified AAV was thawed on ice and the MEF culture media was aspirated. Viral supernatant with a titer varying from 106-1014 viral genome/mL was added to each well and incubated overnight at 37 °C. The viral supernatant was removed after 48 h, replaced with 2 mL of MEF culture medium, and then incubated at 37 °C until GFP expression, about 5 days after transduction. GFP-expressing MEFs were subcultured and validated. Note that for this vector we found that 1012 viral genomes/mL (equivalent to 1010 transducing units/mL) could efficiently transform primary MEF cells to tumorigenic MEFs (Figure 3A). The transduced cells expressed Cre, Cas9, and GFP (Figure 3B,C). To access the tumorigenic potential of the AAV transformed MEF cells, 0.5 x 106 cells (primary/AAV-transduced MEF) were injected into the tongue, lip, and subcutaneously in NOD-SCID mice. Transformed cells formed tumors in these mice, compared with the primary MEF, which did not form tumors (Figure 3D-F).
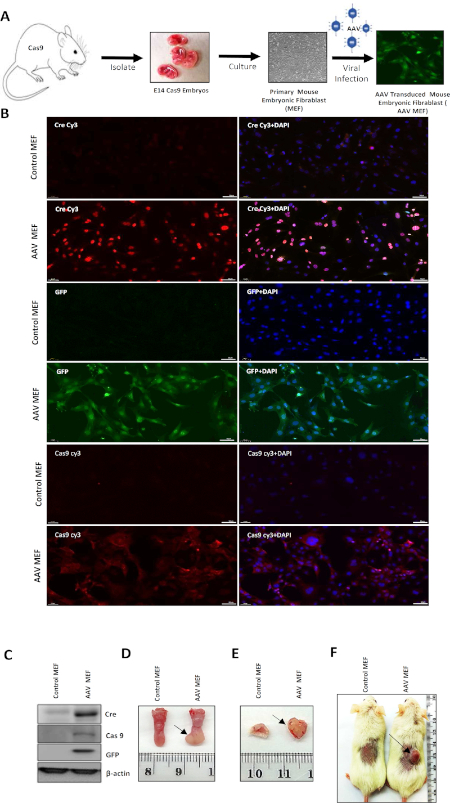
Figure 3: Validation and in vitro transformation of primary MEF cells using the AAV-Cas9 system. (A) Outline of the in vitro transformation of fibroblasts from embryos collected from Cas9 mice using AAV particles. (B) Representative immunofluorescence image of primary MEF and AAV-transduced MEF cells showing the expression of GFP, Cre, and Cas9. DAPI was used to stain the nucleus. (C) Western blotting showing Cre, Cas9, GFP, and β-actin in embryonic normal fibroblasts and genetically edited fibroblasts. (D) Representative image of a tongue tumor that developed in an NOD-SCID mouse after injecting it with transformed fibroblast cells. (E) Representative image of a lip tumor that formed in an NOD-SCID mouse after injecting it with transformed fibroblast cells. (F) Representative image of a flank tumor that developed in an NOD-SCID mouse after injecting it with transformed fibroblast cells. The arrows indicate a tumor. Please click here to view a larger version of this figure.
In vitro generation of the tumorigenic murine tongue epithelial cell line from primary tongue epithelial cells using the AAV-Cas9 system
After validating the transforming property of the AAV system using the MEF cells, we isolated primary tongue epithelial cells from Cas9 mice and cultured them in vitro. The primary tongue cells were transduced with AAV viral particles (1010 cfu/mL), and after one week cells started to express GFP. The tumorigenic transformation of primary tongue epithelial cells (Figure 4A) was confirmed using immunofluorescence and Western blotting (Figure 4B,C). The transformed cells exhibit enhanced KRT 14, E-cadherin, and GFP expression (Figure 4B). The tumorigenic transformation of primary tongue epithelial cells was confirmed by the expression of GFP (i.e., the Cre-dependent expression of Cas9 and GFP) (Figure 4C). The expression of KRT14 and E-cadherin confirms their epithelial cell characteristics. Protein validation confirmed enhanced β-catenin, phospho-ERK, and reduced p53 expression (Figure 4C), indicating the effective downregulation of APC and p53 and the incorporation of mutant KRAS into the tongue cells, thus making them tumorigenic. Genomic analysis by deep sequencing of the DNA isolated from the transformed cells confirmed the effective gene editing and frame-shift of the TP53 and APC genes by the AAV-Cas9 system (Table 2). The tumorigenic property of the AAV-transformed tongue cell line was further assessed by injecting it into the tongue of immunocompetent wild type C57BL/6 mice. The transformed tongue cells, designated as KRAS mutant epithelial cells of the tongue (KRAS Mut EpT), form tumors efficiently in C57BL/6 mice (Figure 4D). KRAS Mut EpT tumors were evaluated by an oral and maxillofacial pathologist by routine hematoxylin and eosin (H&E) staining and by immunohistochemistry. The tumors exhibited a spindle cell morphology with prominent nuclear atypia: pleomorphism, hyperchromatism, giant nuclei, several nucleoli, and a high mitotic rate with atypical mitotic figures. Perivascular invasion and angioinvasion were also noted. These morphological features were accompanied by very limited to nonexistent tumor-associated inflammation. Immunohistochemically, the neoplastic cells were cytoplasmically positive for E-cadherin and KRT 14, both markers of epithelial tissue. KRT 14 is typical of squamous epithelium, supporting the squamous origin of the tumor cells and thus the diagnosis of squamous cell carcinoma (Figure 4E). Hence, this approach provides a versatile methodology for transforming primary cells in vitro with a desired gene alteration. In addition, this methodology facilitates the use of these developed cell lines in vivo in syngeneic mice for better understanding of neoplasia in a real tumor microenvironment milieu.
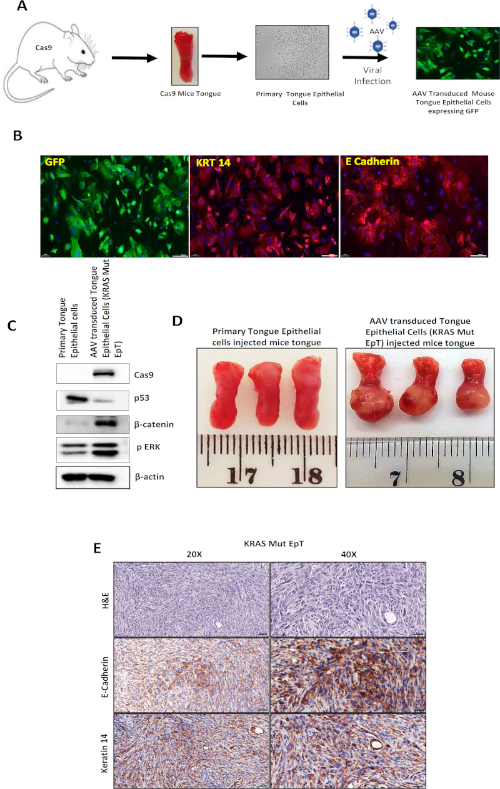
Figure 4: In vitro transformation of primary tongue epithelial cells using the AAV-Cas9 system. (A) Schematic representation of the in vitro transformation of normal epithelial cells obtained from the tongue of Cas9 mice. (B) Representative immunofluorescence image of AAV-transduced tongue epithelial cells showing the expression of KRT14, E-cadherin, and GFP. (C) Western blot showing Cas9, β-catenin, and phospho-ERK upregulation with p53 protein level downregulation, showing the genetically edited epithelial cells. (D) Representative image of a tumor that developed in the tongue of the immunocompetent mice after injecting them with transformed tongue epithelial cells (KRAS Mut EpT). (E) Representative images of tumor tissue sections for H&E, E-cadherin, and KRT 14 staining at 20x and 40x magnification. Please click here to view a larger version of this figure.
| Cell lysis buffer for viral harvesting | Volume |
| 150 mM NaCl | 876.6mg |
| 50 mM Tris-HCl | 605.7mg |
| Use HCl to adjust pH 8.5 | |
| Molecular grade water | Make up to 100 mL |
| Equilibration buffer | |
| 1 mM MgCl2 | 9.52 mg |
| 2.5 mM KCl | 18.64mg |
| Mix all in PBS 1X and adjust pH to 7.2 | Make up to 100 mL |
| Elution buffer | |
| 0.5 M NaCl | 2.92g |
| Mix all in PBS 1X and adjust pH to 7.2 | Make up to 100 mL |
| 10X Triple Enzyme Stock Solution: | |
| Collagenase | 1 g final conc. [10 mg/ml] |
| Hyaluronidase | 100 mg [1 mg/ml] |
| DNase | 20,000 Units final conc. [200 mg/ml] |
| PBS 1X | Make up to 100 mL |
| Plasmid mix (for one 14.5cm Plate) | |
| AAV pCM109 EFS Cre sg APC sg KRAS sg P53- KRAS HDR | 10µg |
| AAV 2/9 capsid vector | 10µg |
| pAD Delta F5 helper | 10µg |
| PEI (1µg/µl Stock) | 90µl |
| DMEM | 1 mL |
Table 1: Recipes of buffers used in this study.
Table 2: Genome analysis data of Kras Mut EpT cells. Please click here to view this table (Right click to download).
