After injecting 1000 TB32048 cells into the pancreas, orthotopic tumors take approximately 30 days to develop (Figure 1A,B). Basement membrane leakage during surgery can cause large tumors to form directly on the peritoneal wall, which are prominently visible through the skin (Figure 1C). We would remove these mice from the study. However, with good surgical skills the incidence of leakage is minimized. Orthotopic tumors harvested at endpoint can grow to a substantial size in C57BL/6 wild-type mice (Figure 1D). Harvested orthotopic tumors require digestion in collagenase/ DNase for 20 min in order to achieve a single-cell suspension (Figure 2). At this point, tumor-derived cells can be plated in a U-bottomed plate at 2 x 106 cells/well. The number of cells plated can be altered depending on the prevalence of T cells within the sample; the cell number can be lowered if T cells are at a high density. Control spleen or lymph node samples can also be plated at this point for stimulation. Each well is stimulated with PMA and ionomycin for 5 h and after 1 h incubation, brefeldin A and monensin are added in order to block extracellular release of cytokines (Figure 2). After the incubation, samples are stained for extracellular epitopes and intracellular cytokines for analysis by flow cytometry (Figure 2).
Samples of spleen and tumors from mice bearing orthotopic tumors were analyzed by flow cytometry. The gating strategy used in flow cytometry analysis for the spleen and orthotopic tumors excludes debris using FSC-A, SSC-A, doublets by FSC-A/FSC-H and SSC-A/SSC-W, then dead or apoptotic cells as positive for fixable viability dye (Figure 3A). Immune cells are then gated on as CD45+, and T cells further gated on as CD3+ from which CD4+ and CD8+ subsets are defined (Figure 3A). A fluorescence minus one (FMO) is performed to determine background fluorescence for gating and a brefeldin A/monensin only control is performed to determine basal production of cytokines (Figure 3B-D).
For IFNγ, incubation with brefeldin A/ monensin resulted in no increase in IFNγ over FMO control in both spleen and tumor samples. However, the addition of PMA and ionomycin increased the% of intracellular IFNɣ detectable in both splenic and tumor-derived CD4+ and CD8+ T cells.
Splenic CD4+ and CD8+ T cells, used as a positive control, have a relatively higher IFNγ production than tumor-infiltrating T cell subsets, with an average of 6.60 ± 1.5 % and 12.97 ± 3.4 % compared to 4.81 ± 1.0 % and 4.13 ± 1.3 %, indicating immunosuppression occurs within the tumor (Figure 3B and 4A). Using the same strategy for TNFα, we visualized that a high percentage of splenic CD4+ T cells are positive for intracellular TNFα (65.93 ± 2.3%), compared to tumor-infiltrating CD4+ T cells (22.45 ±5.4%). Splenic and tumor-infiltrating CD8+ T cells produce similar levels of TNFα (25.15 ± 3.7 % and 19.91 ± 5.1 %, respectively) (Figure 3C, Figure 4B).
Finally, CD107a is an endosomal marker that is expressed transiently on the cell surface during the exocytosis of cytotoxic granules and cytokines, as such, it is used as a surrogate marker for cytotoxicity. The benefit of staining for CD107a during the stimulation is that all transiently cell-surface expressed CD107a will be captured by the fluorescent-antibody. The basal levels of CD107a are shown in brefeldin A/monensin only treated cells. For splenic CD8+ T cells, stimulation with PMA/ ionomycin increases the level of CD107a detected, with the strongest upregulation in CD8+ cells which were 23.95 ± 3.5% CD107a+, compared to 5.8 ± 1.9 % in tumor-infiltrating CD8+ cells, indicating splenic CD8+ had a greater rate of degranulation. On the other hand, splenic and tumor-infiltrating CD4+ expressed comparable levels of CD107a 9.37 ± 1.5 % and 11.50 ± 1.8 % (Figure 3D and 4C).
Overall these results highlight that orthotopic tumors can be generated from the injection of a very low number (1,000) of tumor cells into the pancreas. These tumors can be rapidly digested for the isolation of T cells for ex vivo stimulation. Detection of intracellular cytokines is possible and highlights the basal level of immunosuppression of infiltrating T cells, compared to T cells in secondary lymphoid organs.
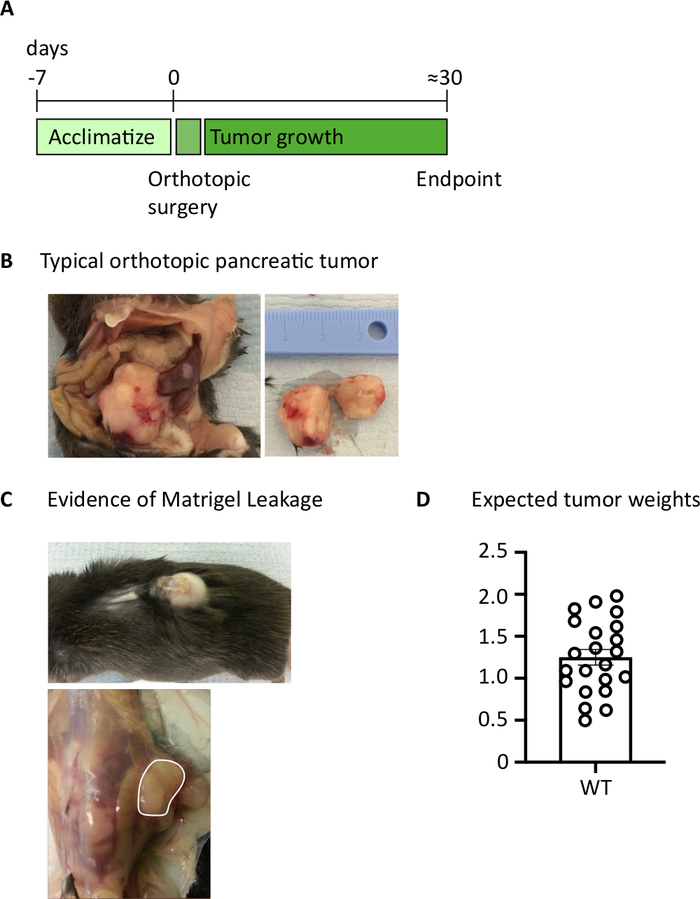
Figure 1: Generation of orthotopic pancreatic tumors. (A) Schedule of in vivo experiments. (B) The macroscopic appearance of orthotopic tumors within the abdominal cavity (left) and after excision (right) where the tumor shown has been cut in half. (C) Evidence of basement membrane leakage during surgery can cause tumors to develop which are visible through the skin (upper photo) and form on the peritoneal wall (lower photo). (D) Orthotopic pancreatic tumor weights harvested from mice which had reached endpoint (n=22). Each data point represents an individual mouse, bar graph shows mean ± SEM. The data in this figure has been modified from previously published work10. Please click here to view a larger version of this figure.
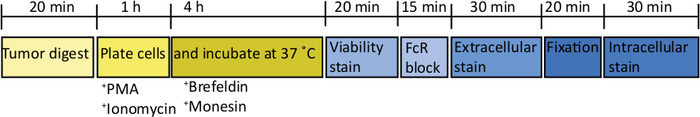
Figure 2: Schematic of processing orthotopic tumors for ex vivo T cell stimulation. After harvesting, pancreatic tumors are rapidly digested in Collagenase (2 mg/mL) and DNase (0.025 mg/mL) for 20 min at 37 °C. Following this, cells are resuspended at 2 x 106/mL in complete RPMI media and plated in a U-bottomed plate. A stimulation cocktail of PMA and ionomycin is added for 5 h, at which point the anti-mouse CD107a antibody can also be added to the culture. After 1 h incubation the intracellular transport blockers, brefeldin A and monensin, are added. After ex vivo stimulation the cells are transferred into a v-bottomed plate for staining with the fixable viability dye (in PBS) for 20 min 4 °C. Cells are washed in FACS buffer and incubated in anti-CD16/32 (FcR block) for 15 min (in FACS buffer) and then incubated with extracellular fluorescent-conjugated antibodies for a further 30 min (in FACS buffer). Cells are washed again in FACS buffer and resuspend in intracellular fixation buffer for 20 min. After this, cells are washed once in FACS buffer and once in 1x permeabilization buffer. Cells are resuspended for 1 h at RT in intracellular fluorescent-conjugated antibodies for 1 h (in 1x permeabilization buffer). Cells are washed once in 1x permeabilization buffer and once in FACS buffer before resuspending in FACS buffer for acquisition on the flow cytometer within 24 h. Please click here to view a larger version of this figure.
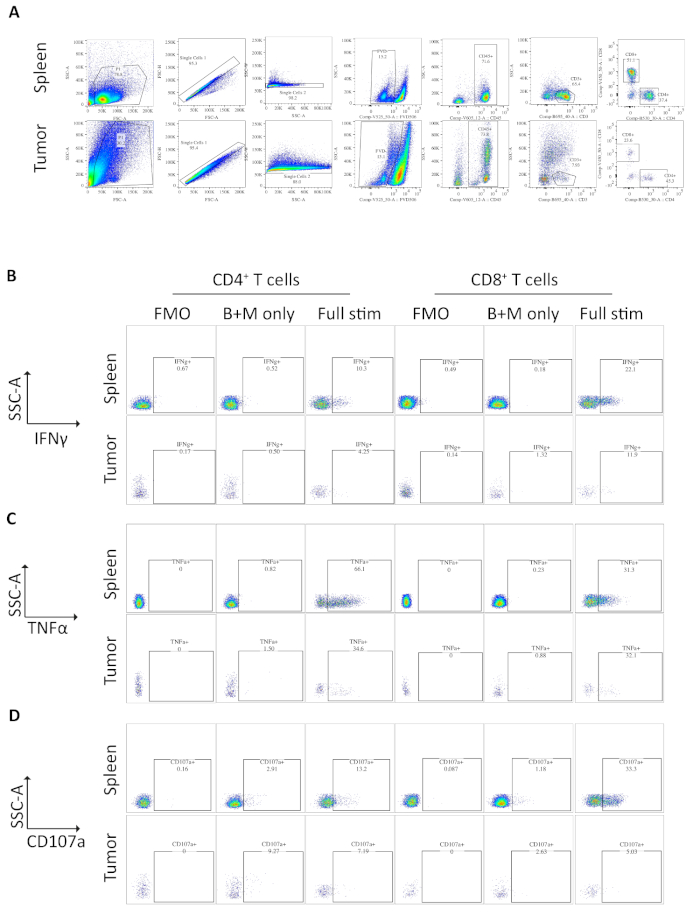
Figure 3: Flow cytometry analysis of ex vivo stimulated spleen- and tumor-derived T cells. (A) Flow cytometry gating strategy used for spleen (positive control) and orthotopic tumors samples. Cells are discriminated from debris using FSC-A/SSC-A and single cells are further isolated using FSC-A/FSC-H and SSC-A/SSC-W. Dead or apoptotic cells are excluded using the fixable viability dye -FVD506 and immune cells are gated on by CD45+. Following this CD3+ T cells and CD4+ and CD8+ subsets are defined. Data was acquired on a BD Fortessa. (B) The gating strategy used to quantify IFNγ+ CD4+ and CD8+ T cells. A fluorescent minus one (FMO) control is used on fully stimulated samples (PMA/ionomycin/brefeldin A/monensin) to determine the background fluorescence. A brefeldin A/monensin only control (B+M only) is used to determine the basal cytokine production. The fully stimulated sample is then used to calculate the % IFNγ+ T cells. (C) The gating strategy used to quantify TNFα+ CD4+ and CD8+ T cells. An FMO control is used on fully stimulated samples (PMA/ionomycin/brefeldin A/monensin) to determine the background fluorescence. A brefeldin A/monensin only control (B+M only) is used to determine the basal cytokine production. The fully stimulated sample is then used to calculate the % TNFα+ T cells. (D) The gating strategy used to define CD107a+ CD4+ and CD8+ T cells. An FMO control is used on fully stimulated samples (PMA/ionomycin/brefeldin A/monensin) to determine the background fluorescence. A brefeldin A/monensin only control (B+M only) is used to determine the basal degranulation. The fully stimulated sample is then used to calculate the % CD107a+ T cells. All flow cytometry data was analyzed on FlowJo Version 10.6.1. The data in this figure has been modified from previously published work10. Please click here to view a larger version of this figure.
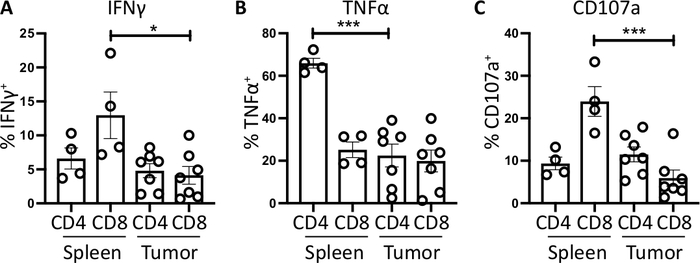
Figure 4: Quantification of ex vivo spleen- and tumor-derived T cell activity. The proportion of CD4+ and CD8+ T cells positive for (A) IFNγ+ (B) TNFα+ and (C) CD107a+ was quantified in the spleen (n=4) and tumor (n=7) of orthotopic-tumor bearing mice. Each data point represents an individual mouse and error bars display mean ± SEM. Statistical significance was tested using an unpaired t-test where * = p<0.05 and *** = p<0.001. All data was analyzed using Prism 8. The data in this figure has been modified from previously published work10. Please click here to view a larger version of this figure.
