Overview of the strategy for visualizing, quantifying, and mapping cell populations of interest in the TME
To quantify cell populations of interest (COIs) in different tissue compartments (TCs) and to characterize their spatial organization, we designed a workflow that integrates affordable and easy to use techniques and maximizes the positional information that can be obtained from precious FFPE clinical specimens (Figure 1). First, serial whole tissue FFPE sections were stained for visualization of COIs (e.g., immune cells) and TCs (e.g., stroma versus parenchyma) (Figure 1, step 1). The number of consecutive sections to be stained should be kept to the minimum that allows visualization of the cells of interest or tissue features needed for addressing the research question. The smaller the number of serial sections, the higher the tissue architecture resemblance and concordance across contiguous sections. In addition, the multiplexing capability can be expanded through reuse of fluorescently stained sections through stripping and reprobing techniques19.
Once the staining steps were done, a whole slide scanner was used to digitize the images. Images acquired from serial sections were aligned and consolidated into a virtual multiplex slide in an automated fashion (Figure 1, section 2). Next, an ROI for the tissue was delineated with a user-defined protocol that identified tissue-associated pixels (TAPs) (Figure 1, step 3). Subsequently, the ROI tissue was segmented into TCs defined as additional ROIs. (Figure 1, step 4). Next, user-defined protocols detected and quantified COIs in different TCs (Figure 1, step 5). Finally, tissue heatmaps of COIs were generated based on their densities and their tissue coordinates (Figure 1, step 6).
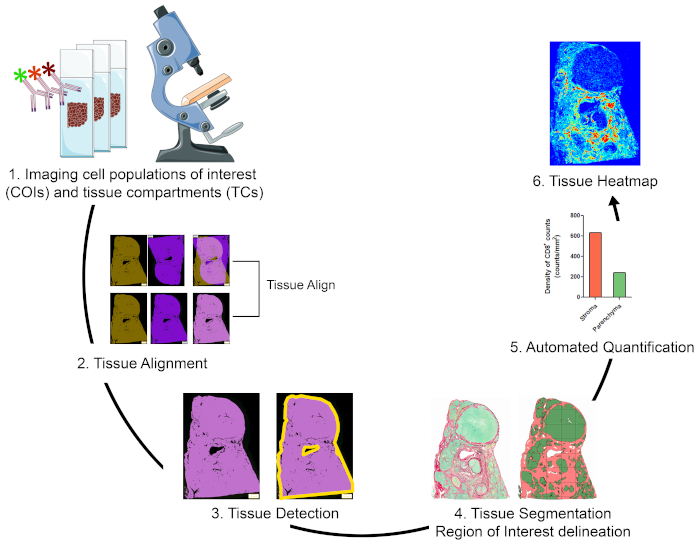
Figure 1: Schematic representation of the strategy for visualizing, quantifying, and mapping immune cells in the TME. (1) Serial whole tissue sections were stained for labeling COIs and TCs. Stained whole tissue sections were digitized using a whole slide scanner. (2) Images acquired from serial sections were linked, aligned, and coregistered in an automated fashion using a Tissuealign analysis module. A composite image was generated from the high-precision alignment of individual images. (3) A user-defined protocol was used for automated detection of tissue-associated pixels (TAPs) in the composite image. (4) The tissue was segmented into TCs (e.g., stroma and parenchyma) defined as ROIs. (5) User defined protocols were used for the automated detection and quantification of COIs in different TCs. (6) Tissue heatmaps of COIs were generated. Please click here to view a larger version of this figure.
Imaging COIs and TCs
Three serial FFPE whole tissue sections of resected tumor from a subject with HBV-associated hepatocellular carcinoma were stained in one or more rounds of staining as in Figure 2A. Section I was stained with H&E to show the tissue architecture, cell morphology, and to determine clinically relevant parameters such as type of malignancy, tumor grade, and overall assessment of immune infiltration (Figure 2C). In contiguous section II, two rounds of mIF were used for labeling liver parenchymal and non-parenchymal cells (Figure 2A). In the first round, normal and tumor vessels were visualized using CD34 staining of endothelial cells. Additionally, epithelial cells (hepatocytes and cholangiocytes) were identified using cytokeratin 8/18, and fibrogenic activated hepatic stellate cells were identified as alpha smooth muscle actin positive (αSMA+) cells (Figure 2C). Following image acquisition, tissue sections were stripped and reprobed with antibodies against macrophages (CD68), and myofibroblasts (desmin). To better characterize the tumor immune infiltrate, adjacent serial section III was stained using two rounds of mIF for the cellular markers CD3, CD4, CD8, forkhead box P3 (FoxP3), and myeloperoxidase (MPO). In all cases DAPI was used as a nuclear counterstain. Finally, section III was stained with PSR stain and counterstained with fast green to visualize fibrillar collagen and segment the tissue into stroma and parenchyma (Figure 2C).
A whole slide scanner equipped with a 20X objective lens was used to digitize stained sections and to create virtual slides. Six images were acquired from the three serial sections (Figure 2B) and the virtual slides subsequently analyzed using the VIS software according to the schematic representation in Figure 1.
Image Analysis
The image analysis comprised five steps: 1) tissue alignment; 2) tissue detection; 3) tissue segmentation; 4) automated quantification of COIs; and 5) tissue heat mapping. All protocols for image analysis were developed using the Author module of the image analysis software and are referred to in the text as APP.
Tissue alignment
Six virtual slides from three serial sections, spanning 11 markers plus H&E and PSR stains, were loaded into the Tissualign module of the image analysis software. Next, the images were linked, aligned, and coregistered in an automated fashion, generating an 11-plex plus H&E and PSR virtual composite image, containing all the layers of the individual images (Figures 2A–C). Alignment was accurate in the case of images originating from adjacent serial sections, showing corresponding tissue structures positioned and arranged in a homologous fashion upon alignment (Figure 2C and Figure S1A). Furthermore, the alignment was precise at the individual cell level for images originating from the same section (Figure S1B). The time for automatic alignment depends on the number, size, complexity, and similarity of the images to be aligned. The alignment of the above-mentioned six virtual slides took 15 min in our VIS station.
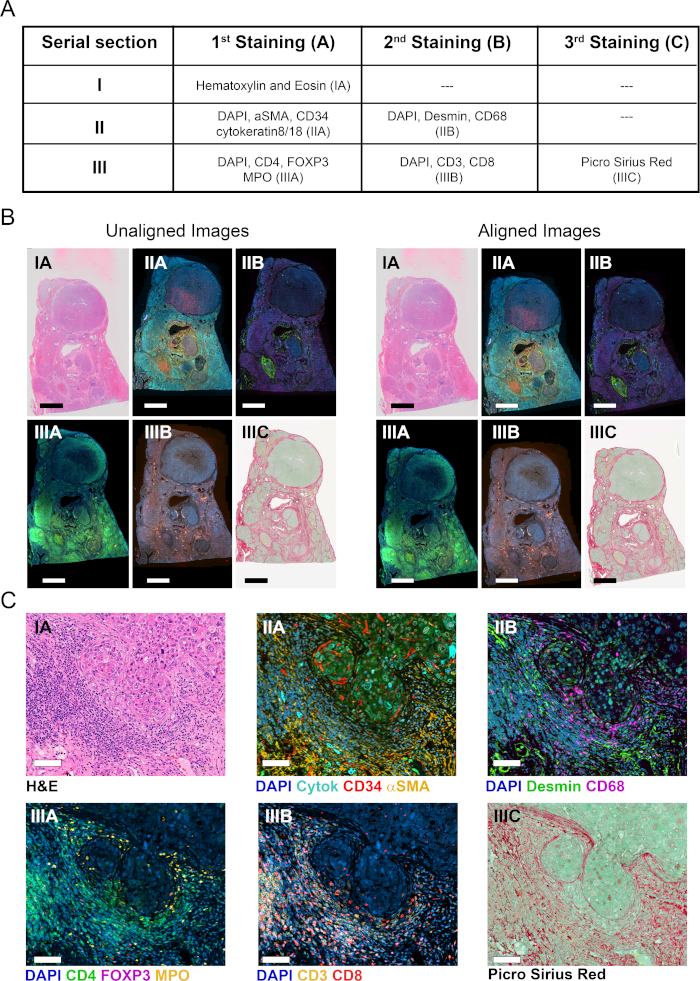
Figure 2: Staining of serial tissue sections and image alignment. (A) Summary of stainings done on three serial sections for visualization of COIs and TCs. Numbers in brackets indicate image designation. For sections II and III, tissues were stripped and reprobed with a second cocktail of antibodies. (B) Overview of six individual whole tissue images before and after tissue alignment (left and right, respectively). Scale bar = 3,500 µm. (C) Zoomed view of aligned images. Scale bar = 80 µm. Please click here to view a larger version of this figure.
Tissue Detection
Once the images were linked and aligned, we sought to identify the TAPs (Figure 3A). To design an APP for the automated detection of TAPs (APP 1, Table 1), we took advantage of two properties that differentiate TAPs from pixels not associated with tissue. First, the DAPI signal (blue band) is restricted to the nuclei, which are located exclusively in the tissue, meaning that all DAPI+ pixels are a subset of TAPs. Second, TAPs have higher autofluorescence signal in the green and yellow bands compared to pixels not associated with the tissue. Consequently, we developed APP 1 for tissue detection (Table 1), which detects the TAPs based on baseline signal in these channels using simple thresholding techniques. Thresholds for the blue, green, and yellow bands were set so that TAPs had background intensity values above the thresholds, while pixels not associated with the tissue had values below. APP 1 for tissue detection was applied to image IIA, which contains layers in the blue, green, and yellow channels (Figure 3A). As outputs of APP 1, a bright green mask was laid down on top of the TAPs, and a ROI called "Tissue" was delineated (output, Figure 3A). Furthermore, the area of the tissue was determined as a quantitative output variable. Because APP 1 does not incorporate the pixels not associated with the tissue into the ROI Tissue, they were excluded from subsequent analysis based on this ROI (Figure 3A). The precision of APP 1 at identifying TAPs is shown in Figure 3A.
Tissue segmentation and delineation of ROIs for TCs
Next, we proceeded to define different compartments inside the ROI tissue by segmenting the tissue into stroma versus parenchyma. We used the PSR stained image (IIIC, Figure 2C), where the stroma can be defined as the area associated with the deposition of fibrillar collagens (red band), the parenchyma as the area where fibrillar collagens are absent, and the fast green counterstaining dye prevails (green band) (Figure 3B). We created APP 2 (Table 1) to digitally delimit the TCs Stroma and Parenchyma. This APP works on the predefined ROI Tissue (output, Figure 3A) and uses representative stroma and parenchyma areas for training the Classifier tool integrated in the Image Analysis module. The trained Classifier assigns the pixels to either a stroma or a parenchyma label (salmon and green, respectively, Figure 3B). Upon classification of pixels, APP 2 executed morphological operations aiming at defining the ROIs Stroma and Parenchyma (Figure 3B and Table 1). The performance of APP 2 at classifying pixels and generating the respective ROIs is shown in Figure 3B. Additionally, APP 2 quantifies the area of the stroma and the parenchyma. Finally, even though the segmentation is done using the PSR stained section, the outlined stroma and parenchyma regions can be transferred to any image aligned to the PSR image.
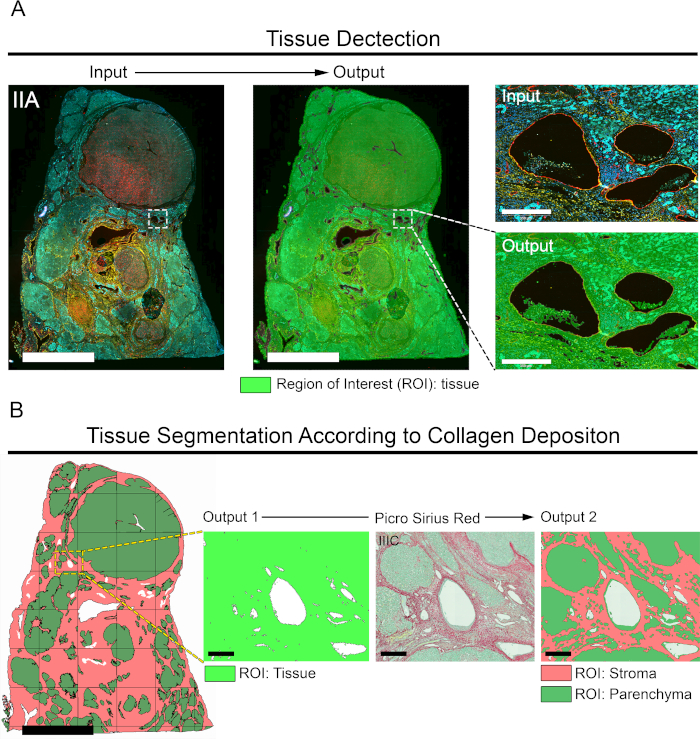
Figure 3: Automated tissue detection/segmentation and generation of respective ROIs. (A) Image IIA was used to identify the TAPs (left image, scale bar = 6,000 µm). A bright green mask was assigned to the TAPs using APP 1 (Table 1) generating a ROI called Tissue (output 1). Right, inset shows zoomed view demonstrating the precision of APP 1 at detecting TAPs. Scale bar = 350 µm. (B) The ROI Tissue (output 1) is segmented into stroma and parenchyma using APP 2. The image on the left shows a view of the ROI Tissue segmented into ROI stroma (salmon) and ROI parenchyma (green). Scale bar = 4,500 μm. On the right, zoomed views of inset for ROI Tissue, the original PSR staining (image IIIC), and the ROIs stroma and parenchyma. Scale bar = 250 μm. Please click here to view a larger version of this figure.
Automated quantification of COIs
Next, we proceeded to identify, locate, and quantify COIs in the ROIs Stroma and Parenchyma. APPs 3 to 8 (Table 1) were created to locate and count the following COIs: CD4+FoxP3+, CD8+, CD68+, MPO+, αSMA+, and CD34+ cells, respectively. APP 3 was designed to locate and count CD4+FoxP3+ cells (image IIIA, Figure 2C) as surrogate markers of regulatory T cells (Tregs). This protocol detects colocalization of the signal from the nuclear transcription factor FoxP3 (red band) and the DNA labeling dye DAPI (blue band). Given that recently activated T cells upregulate FoxP3, to enrich for Tregs we set thresholds for preselecting only bright FoxP3+ cells (FoxP3hi). Next, out of all preselected DAPI+FoxP3hi cells, only those that were surrounded by bright ring-shaped CD4 signals (green band) were labelled and counted as FoxP3hiCD4+ cells (pink label, Figure 4A). The density of FoxP3hiCD4+ cells in the ROIs Stroma and Parenchyma were determined as quantitative output variables of APP 3 (Figure 4A).
Similarly, APPs 4 to 6 were designed for the detection of CD8+, CD68+, and MPO+ cells. These APPs share the same baseline design for detecting and quantifying COIs. Specifically, COIs are identified based on signal intensity from the specific cell population biomarker, and then several postprocessing morphological steps are executed to delineate individual cells (Table 1). The individual cells or COIs are labelled, counted, and their tissue coordinates registered. APPs 4 to 6 also determine the density of the COIs in the ROIs Stroma and Parenchyma (Figure 4B–D).
The quality of our DAPI staining was not good enough for integrating nuclei segmentation into APPs 3 to 6, so we cannot ensure that all individually labelled objects are individual cells. For this reason, we expressed the density of cells in counts of labeled objects/mm2 (Figure 4). However, cell aggregates were successfully separated into individual cells in the postprocessing steps built into APPs 3 to 6, and extensive visual inspection showed that most labeled objects corresponded to single cells.
For detecting αSMA+ and CD34+ area, we developed APPs 7 and 8, respectively (Table 1). Both APPs detect the specific signal based on thresholds and determine the percentage of positive area in the ROIs Stroma and Parenchyma (Figure 4E–F).
One of the most interesting possibilities of generating virtual multiplex slides is the analysis of colocalization expression. We generated APP 10 to detect colocalization between αSMA and desmin, two markers co-expressed by myofibroblasts in the liver. APP 10 uses thresholds for finding pixels positive for αSMA, desmin, and αSMA plus desmin (Table 1). As quantitative output variables, APP 10 determines the αSMA+ area, the desmin+ area, and the area of colocalized expression of these two markers (Figure S3).
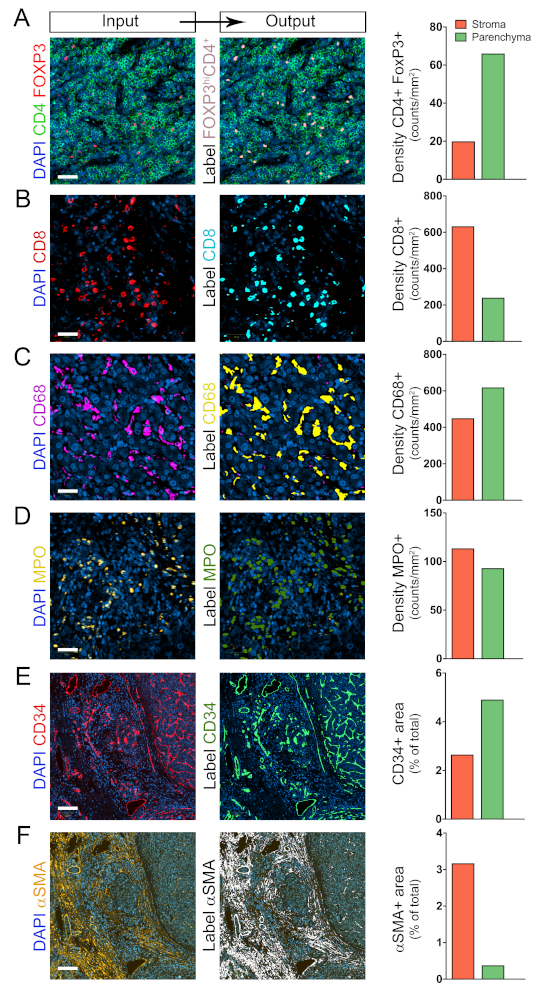
Figure 4: Identification and quantification of COIs in the TCs stroma and parenchyma. (A–F) Automated detection and quantification of CD4+FoxP3+, CD8+, CD68+, MPO+, αSMA+, and CD34+ COIs in the ROIs Stroma and Parenchyma using protocols 3, 4, 5, 6, 7, and 8, respectively (Table 1). Shown on the left are the original images, in the middle the processed images, and on the right the quantifications. For Figures 4A–D, scale bar = 40 µm. For Figures 4E and F, scale bar = 350 µm. Please click here to view a larger version of this figure.
As an alternative to quantifying the COIs in the TCs Stroma and Parenchyma, we determined the density of immune cells in the different malignant nodules named 1 to 4 (Figure 5A, H, and I). The ROI for each nodule was manually delineated as indicated in Figure 5A. Distinctive tissue immune signatures characterized each nodule, further revealing the intrinsic heterogeneity of the TME.
Tissue Heatmaps
As mentioned above, APPs 3 to 8 store the tissue coordinates of every individually labelled object. This feature allows the automated generation of tissue maps where regions of high density of a given cell population are displayed as hot spots (red), and regions with relatively low density as cold spots (dark blue). Intermediate density values are assigned colors according to the color scale shown in Figure 5. Tissue heatmaps were generated by APPs that divided the images into circles of 50 μm diameter and assigned a color according to the relative density of a given COI inside the circle. As displayed in Figure 5B–G, the positioning patterns and intensity distribution of the different COIs in the TME was quite varied. Furthermore, at the level of individual nodules, the arrangement of different populations in the tissue area was unique (Figure S2A–C). To provide an example of the power of this technique and to visualize the spatial organization of hot spots from different populations in the same nodule, the hot spots from individual cell types were manually extracted and mapped together onto the outline of nodule 2 (Figure S2, Figure D, and Figure E).
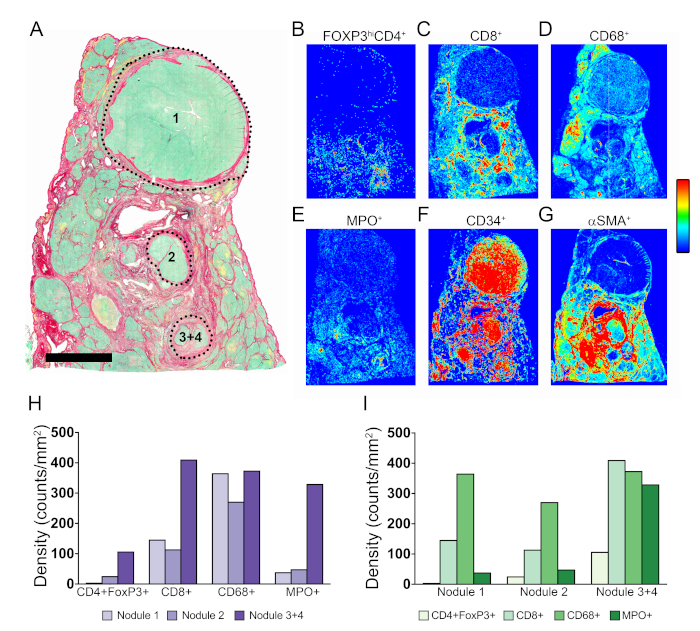
Figure 5: Tissue heatmaps of COIs in the TME. (A) Picrosirius Red staining showing location of nodules 1, 2, 3, and 4. (B–G) Tissue heatmaps for CD4+FoxP3+, CD8+, CD68+, MPO+, CD34+, and αSMA+ COIs, respectively. Dark blue indicates relative low density, and red indicates relative high density. Intermediate density values are assigned colors according to the shown color scale. (H and I) Quantification of COIs in nodules 1, 2, and 3 + 4 organized per cell type and per nodule, respectively. Please click here to view a larger version of this figure.
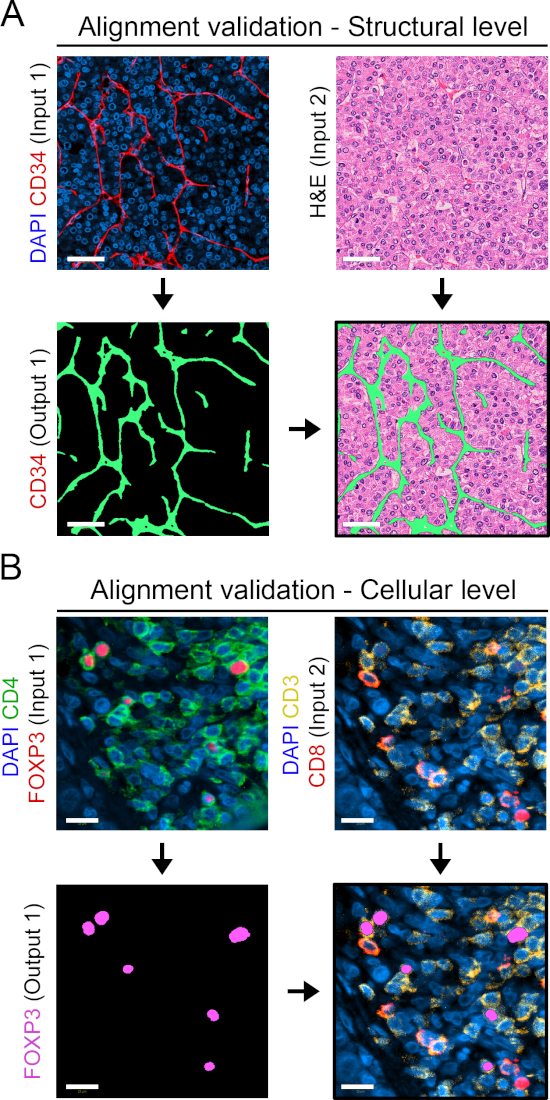
Supplementary Figure S1: Validation of tissue alignment. (A) CD34 staining (in red) done on section II (input 1) is used for generating a CD34 mask in green (output 1). The green mask (output 1) is overlaid on the H&E image from the aligned serial section I (input 2). The merge image shows perfect correspondence of vascular structures. Scale bar = 50 μm. (B) Image IIIA showing the merge of DAPI, CD4, and FoxP3 (input 1) was used to generate a label for CD4+FoxP3+ cells (output 1 in magenta). Output 1 label was transferred onto aligned image IIIB (input 2) and shows perfect correspondence between the pairs FoxP3/DAPI, and CD4/CD3 in the merge image. Scale bar = 15 μm. Please click here to view a larger version of this figure.
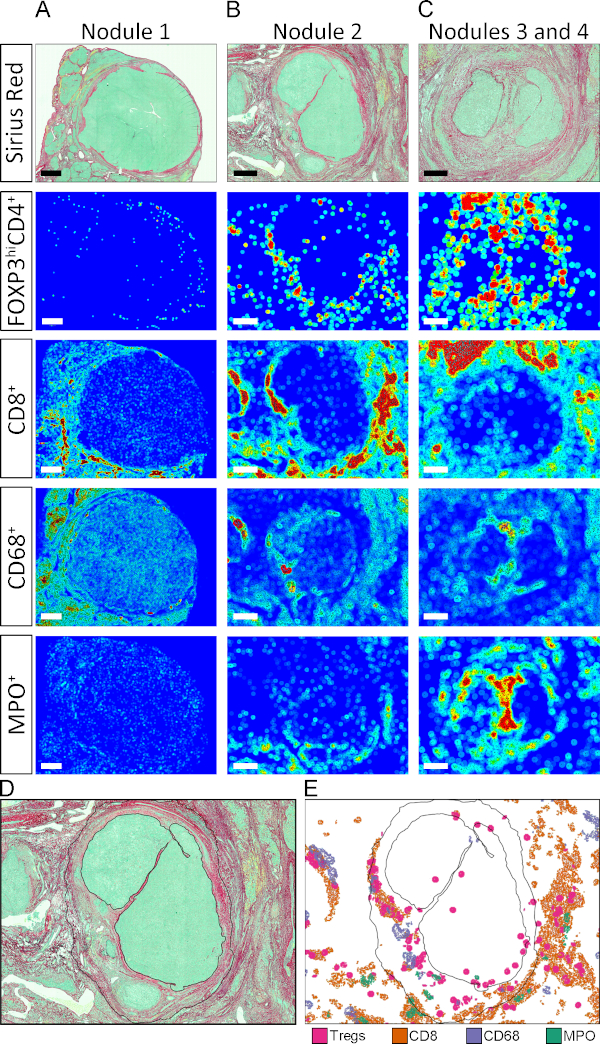
Supplementary Figure S2: Zoomed view of tissue heatmaps. (A–C) Tissue heatmaps for CD4+FoxP3+, CD8+, CD68+, and MPO+ cells in nodules 1–4. Scale bars in nodules 1, 2, and 3 + 4 represent 1,500 μm, 700 μm, and 500 μm respectively. (D) Outline of nodule 2 with black solid line. (E) Hot spots for CD4+FoxP3+, CD8+, CD68+, and MPO+ cells in nodule 2 were extracted and mapped together onto the nodule 2 outline defined in D. Please click here to view a larger version of this figure.
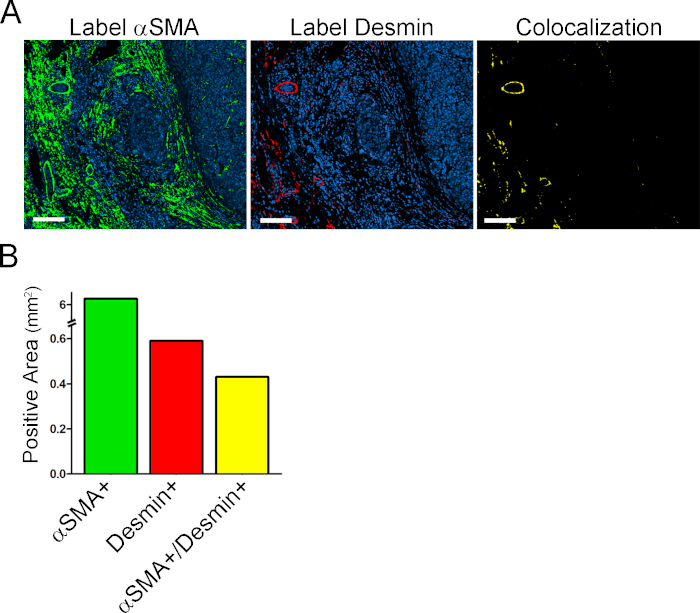
Supplementary Figure S3: Colocalization Analysis. (A) On the left and middle are images of αSMA label in green and desmin label in red respectively. On the right is a αSMA/desmin double positive area in yellow. (B) Quantification of αSMA+ area, desmin + area, and αSMA/desmin double positive area. Scale bar = 150 μm. Please click here to view a larger version of this figure.
| APP | Purpose | Classification | Classification | Post-Processing Steps | Output Variables |
| Method | Features | ||||
| (pixel value) | |||||
| 1 | Tissue detection | Threshold | Channel DAPI (150) | o Label objects with colocalized above-threshold values for the 3 channels | o ROI Tissue |
| Channel FITC/A488 (120) | o Close positive object 5 pixels | o Tissue Area | |||
| Channel TRITC/A568 (40) | o Create ROI Tissue | ||||
| 2 | Tissue Segmentation | Decision Forest | RGB-R median | o Fill holes | o ROI Stroma |
| RGB-G median | o Create ROI Stroma | o Stroma Area | |||
| RGB-B median | o Create ROI Parenchyma | o ROI Parenchyma | |||
| IHS-S median | o Parenchyma Area | ||||
| H&E Eosin median | |||||
| 3 | To locate and quantify CD4+ FoxP3+ cells | Threshold | Channel DAPI (>600) | o Label objects with colocalization of DAPI and Cy5/A647, surrounded by FITC/A488 signal | o Counts and density of CD4+FoxP3+ cells in ROIs Stroma and Parenchyma |
| Channel FITC/A488 poly smoothing (>850) | o Clear objects smaller than 7 μm2 | o Coordinates of individual CD4+FoxP3+ cells | |||
| Channel Cy5/A647(>800) | |||||
| 4 | To locate and quantify CD8+ cells | Threshold | Channel DAPI (<1200) | o Clear positive objects smaller than 15 μm2 | o Counts and density of CD8+ cells in ROIs Stroma and Parenchyma |
| Channel Cy5/A647 median (>80) | o Close positive objects 2 pixels | o Coordinates of individual cells | |||
| o Separate objects | |||||
| 5 | To locate and quantify CD68+ cells | Threshold | Channel FITC/A488 (>200) | o Clear positive objects smaller than 20 μm2 | o Counts and density of CD68+ cells in ROIs Stroma and Parenchyma |
| o Dilate positive objects 3 pixels | o Coordinates of individual CD68+ cells | ||||
| o Separate objects | |||||
| 6 | To locate and quantify MPO+ cells | Threshold | Channel DAPI (>400) | o Clear objects smaller than 5 μm2 | o Counts and density of MPO+ cells in ROIs Stroma and Parenchyma. |
| Channel TRITC/A568 (900-4000) | o Dilate 3 pixels positive objects | o Coordinates of individual MPO+ cells. | |||
| o Separate objects | |||||
| 7 | To locate and quantify αSMA+ area | Threshold | Channel TRITC/CF568 (>1050) | o Clear positive objects smaller than 25 μm2 | o Counts and density of αSMA+ area in ROIs Stroma and Parenchyma |
| o Dilate 3 pixels positive objects | o Coordinates of αSMA+ pixels | ||||
| 8 | To locate and quantify CD34+ area | Threshold | Channel DAPI (<5000) | o Clear positive objects smaller than 25 μm2 | o Counts and density of CD34+ area in ROIs Stroma and Parenchyma |
| Channel Cy5/A647 median (>120) | o Dilate 3 pixels positive objects | o Coordinates of CD34+ pixels | |||
| 9 | Create tissue heatmaps for a given cell population | Object Heatmap | Object Heatmap | o Heatmap | |
| Drawing radius 50 μm | — | ||||
| 10 | Quantify colocalization between αSMA and Desmin | Threshold | Channel TRITC (CF568) (>1050) | o Label objects with above threshold values for TRITC (CF568) | o Quantify colocalized expression of αSMA and Desmin |
| Channel Cy5 (A647) (>1000) | o Label objects with above threshold values for Cy5 (A647) | ||||
| o Label objects with colocalization of above threshold values for TRITC (CF568) and Cy5 (A647) | |||||
| o Clear positive objects smaller than 25 μm2 |
Table 1: General parameters used for the design of APPs employed for image analysis. The parameters specified in this table are adjusted to the unique characteristics of the images used in this analysis (e.g., background, artifacts, etc.) and may not be applicable to other images. Because the post-processing steps mentioned were defined for the specific images analyzed in this study, they are intentionally not detailed. The user should customize the APPs to the images to be analyzed.
| Section/Staining | Primary Antibody | Secondary Antibody |
| Section II/1st Staining | Mouse IgG2a anti-human αSMA Mouse IgG1 anti-human CD34 Rabbit anti-human Cytokeratin 8/18 |
Goat anti-mouse IgG2a CF568 Rat anti-mouse IgG1 A647 Donkey anti-rabbit A488 |
| Section II/2nd Staining | Rabbit anti-human Desmin Mouse anti-human CD68 |
Donkey anti-rabbit A647 Donkey anti-mouse DyLight 755 |
| Section III/1st Staining | Mouse anti-human CD4 Rabbit anti-human FoxP3 Goat anti-human MPO |
Donkey anti-mouse A488 Donkey anti-rabbit A647 Donkey anti-goat A568 |
| Section III/2nd Staining | Rabbit anti-human CD3 Mouse anti-human CD8 |
Donkey anti-mouse DyLight 755 Donkey anti-rabbit A647 |
Table 2: Primary-Secondary Antibody Pairs for mIF.
