A schematic diagram of the chemotaxis slide used for time-lapse video microscopy of mouse peritoneal macrophages migrating in a chemotactic gradient is shown in Figure 2A. The slide contains three chemotaxis chambers, each of which has four filling ports. Ports can be individually closed using the plugs shown above the slide. Alternatively, a non-sealing cap can be placed over an unplugged port to maintain sterility. After plugging ports 1 and 4, the observation area (1 mm wide x 2 mm long x 70 µm high channel connecting the two reservoirs) between ports 2 and 3 can be prefilled with medium by placing a 15 µL drop into port 3 and aspirating with a 2–20 µL volume pipette at port 2 (Figure 2B). A suspension of mouse resident peritoneal cells (10 x 106 cells/mL) was seeded into the observation area by placing a 10 µL drop of suspension into port 3 and slowly aspirating at port 2 (Figure 2C). A typical image of cells seeded in the observation area taken by phase-contrast microscopy using a 10x objective lens is shown in Figure 2C. After incubating for 2–3 h, the chemotaxis slide was slowly filled with medium (Figure 2D). After plugging ports 1 and 2, medium was slowly injected via port 3 until it emerged from port 4. Next, the plug was switched from port 1 to port 3, and then the second reservoir was filled by slowly injecting medium via port 4 until it emerged at port 1. At this stage, cells in the observation area were reinspected using an inverted microscope (Figure 2E). By comparing images shortly before (Figure 2C) and after (Figure 2E) filling of the reservoirs, up to two-thirds of the cells had been washed out of the observation area. Generally, weakly adherent CD19+ cells (B1 cells) were washed out and the remaining cells were predominantly F4/80+ cells (macrophages). This was demonstrated by fluorescence microscopy after labeling each cell type with fluorescently labeled specific antibodies (Figure 4). In Figure 4A, freshly isolated mouse resident peritoneal cells were labeled with green fluorescent fluorophore-conjugated anti-F4/80 antibodies and red fluorescent fluorophore-conjugated anti-CD19 antibodies, and the nuclei of cells were labeled with a blue fluorescent nucleic acid stain. F4/80 is a specific marker for mouse macrophages30, whereas CD19 is a B cell marker. Figure 4B shows F4/80+ cells imaged by spinning disk confocal microscopy in the observation area of a chemotaxis chamber. The cells were labeled after an overnight chemotaxis assay recorded by time-lapse, phase-contrast microscopy.
Complement C5a (chemoattractant) was introduced to one of the two reservoirs by placing a 15 µL drop of medium containing 0.54 µg/mL (recombinant mouse) complement C5a and 10 µg/mL Patent Blue V into filling port 1 (Figure 3A) after plugging ports 2 and 3. The chemoattractant medium was slowly drawn into the reservoir by slow aspiration with a pipette via port 4. Figure 3B shows the diffusion of the blue dye after drawing the 15 µL drop into a reservoir. Patent Blue V was used as an indirect visual indicator of chemoattractant diffusion. Complement C5a molecules are considerably larger than those of Patent Blue V (9.0 kDa versus 0.57 kDa) and diffuse more slowly. After diffusion of complement C5a in the reservoir, its concentration was ~0.2 µg/mL (15 µL/40 µL [reservoir volume] x 0.54 µg/mL = 0.2 µg/mL), equivalent to ~22.5 nM. A modestly steep gradient formed across the observation area after 3 h and continued to increase, reaching a maximum at around 12 h31. Figure 3C shows the migration tracks of macrophages migrating in a complement C5a gradient, between 6–12 h after adding the chemoattractant. Cell velocity and chemotactic efficiency, indexed as y-FMI (y-forward migration index; range: -1 to +1) and x-FMI, of individual macrophages was calculated from the migration plots (Figure 3D). Figure 3D also shows a migration plot produced after normalizing the start point of each migration track to X = 0 and Y = 0 below the box plots. The inset in the migration plot shows how the y-FMI was calculated for each migration track.
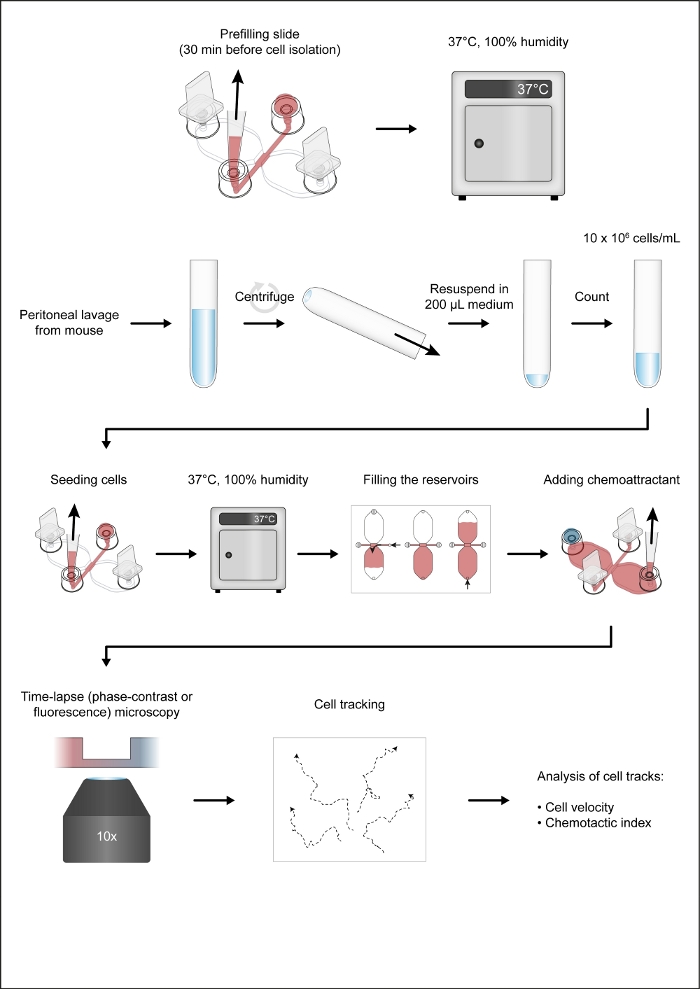
Figure 1: The workflow of the chemotaxis assay. Please click here to view a larger version of this figure.
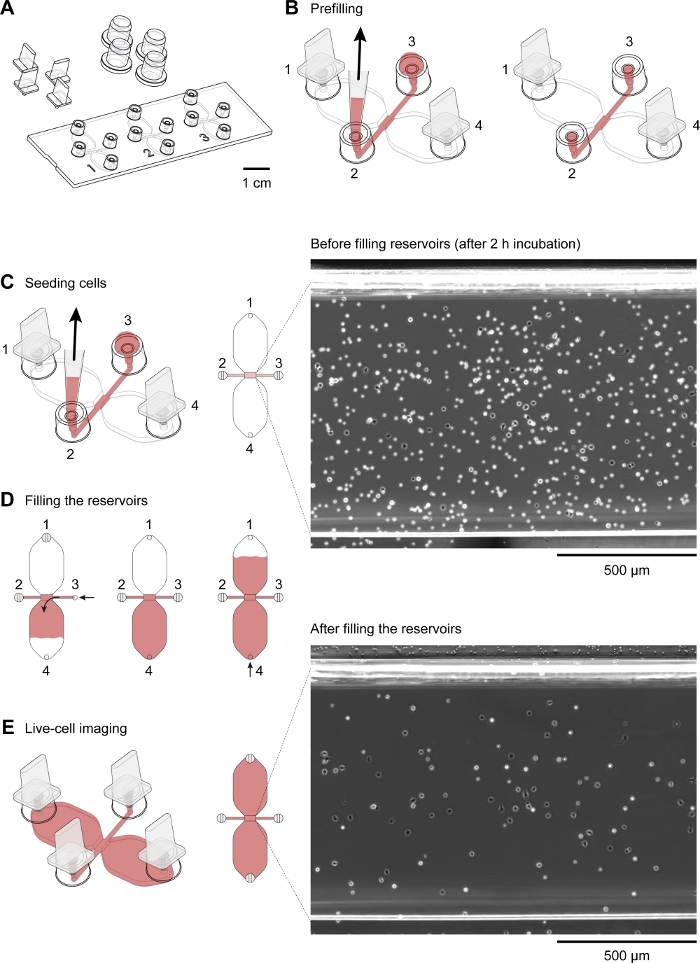
Figure 2: Handling of chemotaxis slides. (A) A 3D view of a chemotaxis slide with four plugs and four caps. The slide contains three chemotaxis chambers, each of which consists of two 40 µL reservoirs connected by a 1 mm x 2 mm channel, which is 70 µm high, termed the observation area. (B) The connecting channel extends at both ends to filling ports 2 and 3. After inserting plugs into filling ports 1 and 4, the observation area was prefilled with medium (red) by applying a drop of medium to port 3 and aspirating at port 2 with a 10–200 µL pipette tip. Subsequently, caps were applied to ports 2 and 3 before incubating the slide at 37 °C and preparing the cell suspension. (C) The observation area, where the chemoattractant gradient formed, was seeded with macrophages by applying a 10 µL drop of mouse resident peritoneal cells at port 3 and slowly aspirating at port 2. The slide was then incubated in a humidity chamber at 37 °C for 2–3 h. The phase-contrast image shown on the right, obtained via a 10x objective lens, shows peritoneal cells after seeding and incubation at 37 °C for 2 h. Scale bar = 500 µm. (D) Chemotaxis chambers were filled with medium by plugging ports 1 and 2 and then slowly injecting medium via port 3 until it emerged at port 4. Slow and steady filling can be achieved by turning the volume setting ring of a 20–100 µL volume pipette. After filling the first reservoir, the second reservoir can be filled by plugging ports 2 and 3 and then slowly injecting medium at port 4 until it emerges at port 1. (E) Phase-contrast image of the same observation area shown above (C) after filling the two reservoirs. Scale bar = 500 µm. Graphic elements provided by Elias Horn. Please click here to view a larger version of this figure.
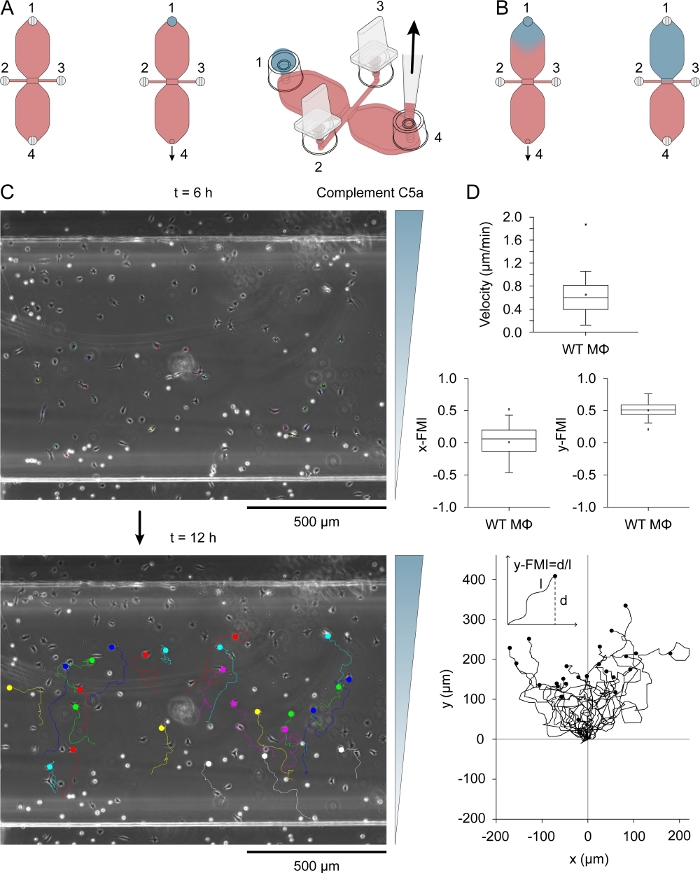
Figure 3: Chemotaxis assay. (A) Chemoattractant was introduced to one of the two reservoirs of a chemotaxis chamber by applying a 15 µL drop of medium containing 0.54 µg/mL complement C5a and 10 µg/mL Patent Blue V to filling port 1, followed by slow aspiration at port 4. (B) Initially after being drawn into the reservoir the blue, chemoattractant-containing medium had a roughly inverted drop shape, and then slowly diffused throughout the reservoir. (C) Migration tracks of macrophages migrating in a chemoattractant (complement C5a) gradient between 6–12 h after introducing chemoattractant to one of the reservoirs. The direction of the gradient is indicated on the right. The end of each migration track is indicated by a filled circle. (D) Blox plots of velocity, x-FMI (x-forward migration index) and y-FMI (y-forward migration index), an index of chemotactic efficiency that ranges from -1 to +1. Data were obtained by analysis of 25 macrophage migration tracks. Macrophages in the lower half of the observation area and showing displacement of at least one cell width over 6 h were randomly selected for analysis. Below is a plot of migration tracks after normalizing the start point to X = 0 and Y = 0. The chemotaxis index (y-FMI) was calculated by dividing the net displacement along the Y-axis (d) by the accumulated length (l) of the migration path, as schematically shown. Graphic elements in panels A and B provided by Elias Horn. Please click here to view a larger version of this figure.
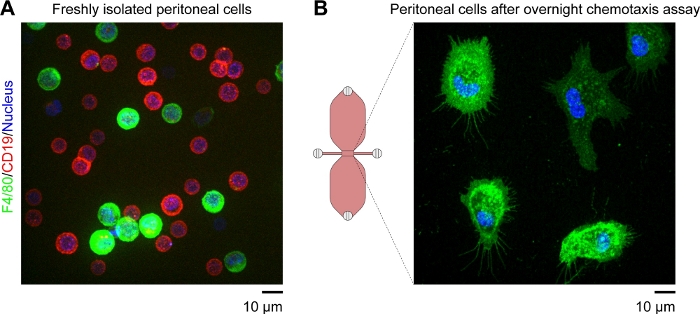
Figure 4: Fluorescent images of living mouse resident peritoneal cells obtained by spinning disk confocal microscopy. (A) Extended focus image (brightest point merge of all Z-planes) of freshly isolated mouse peritoneal cells labeled with green fluorescent anti-F4/80 (macrophage marker) antibodies, red fluorescent anti-CD19 (B cell marker) antibodies, and a blue fluorescent nucleic acid stain. Scale bar = 10 µm. (B) Snapshot (single Z-plane) of F4/80+ cells (macrophages) in the observation area of a chemotaxis chamber taken after an overnight chemotaxis assay. Cells were labeled with green fluorescent anti-F4/80 antibodies and a blue fluorescent nucleic acid stain. The complement C5a and Patent Blue V gradients were washed out by the cell labeling procedure, which explains why the upper reservoir in the schematic diagram of the chemotaxis chamber is not blue. Scale bar = 10 µm. Graphic element provided by Elias Horn. Please click here to view a larger version of this figure.
