In the Lnt reaction the sn-1 fatty acid from phospholipids is transferred onto a diacylglyceryl peptide, resulting in mature triacylated peptide8. The in vitro Lnt assay described here is designed to use phospholipids containing an alkyne fatty acid (alkyne-POPE) and FSL-1-biotin as substrates, resulting in the formation of alkyne-FSL-1-biotin. Upon a click-chemistry reaction with azido-FAM, this product should become fluorescently labeled and detected by fluorescence spectrometry (Figure 2).
The reaction conditions were optimized for maximum fluorescence readout at 520 nm in a plate reader. At 1 ng/µL enzyme, complete conversion of FSL-1-biotin was observed8 (Figure 3). Negative controls included reactions without enzyme, with an inactive variant of the enzyme (active site mutant C387S), or with thiol-specific inhibitor MTSES that result in low fluorescence detection. Biotin-fluorescein bound efficiently to streptavidin-coated plates and was used as an internal control for maximum fluorescence signal. All experiments were performed in triplicate and standard deviation calculations and statistical analysis showed that the assay was sensitive and reproducible.
In order to develop an HTS assay to screen for small molecule inhibitors of Lnt, the quantity of reagents was reduced, and the reactions were performed directly in 384 well plates. Furthermore, wash steps were automated using a plate washer (see Table of Materials). As for the tube assay, the reaction was sensitive and reproducible, with a significant difference between the negative control (C387S) and active enzyme (Lnt) (Figure 4). In HTS the Z’ factor determines whether a response in an assay is large enough for screening purposes9 and is calculated using the following equation:

Where S is the average signal, B is background signal, and σ is standard deviation.
The average Z’ factor was >0.6 for the Lnt assay performed in HTS format using 384 well plates, suggesting that the assay for screening of small molecule for Lnt inhibition was outstanding.

Figure 1: Enzymatic reactions in posttranslational modification of lipoprotein in proteobacteria. The sequential steps were catalyzed by Lgt10, Lsp11, and Lnt12,13,14 in the cytoplasmic membrane. PGN: peptidoglycan, SP: signal peptide, LB: lipobox, conserved motif containing Cys+1 and modified with fatty acids and the first amino acid in mature lipoprotein, PG: phosphatidylglycerol, G-1-P: glycerol-1-phosphate, PE: phosphatidylethanolamine, lysoPE: lyso- phosphatidylethanolamine. Please click here to view a larger version of this figure.

Figure 2: Schematic of the fluorometric enzyme assay for apolipoprotein N-acyltransferase (Lnt). The substrates FSL-1-biotin (yellow) and alkyne-POPE (blue) were mixed with purified Lnt enzyme solubilized in detergent. The reaction was performed in mixed micelles at 37 °C (step 1). The alkyne-FSL-1-biotin product was labeled with fluorescein (orange) by click-chemistry (step 2) and detected in a fluorescence plate reader upon binding to streptavidin-coated plates (step 3). The figure is a modified version of Figure 1B published by Nozeret et al.8 according to the Creative Commons license (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.

Figure 3: Fluorescence detection of Lnt activity in 96 well format. Lnt reactions were performed in tubes at 37 °C for 16 h. After fluorescent labeling by click-chemistry and binding to streptavidin-coated plates, fluorescence was measured by fluorescence spectrometry. Biotin-fluorescein (biotin-fluo 0.26 µM) was used as control for fluorescence. Buffer was reaction buffer only. Samples indicating alkyne-POPE (50 µM), FSL-1-biotin (50 µM), and substrates (mix of alkyne-POPE and FSL-1-biotin, 50 µM each) did not contain enzyme. Negative controls included inhibition with MTSES (10 mM) and an inactive variant of Lnt (C387S). Lnt enzyme was added at 1 ng/μL. Standard deviations were calculated for n = 3 experiments. ** P-value < 0.005. Excitation at 494 nm, bandwidth 5 nm and emission at 520 nm, bandwidth 5 nm. Please click here to view a larger version of this figure.
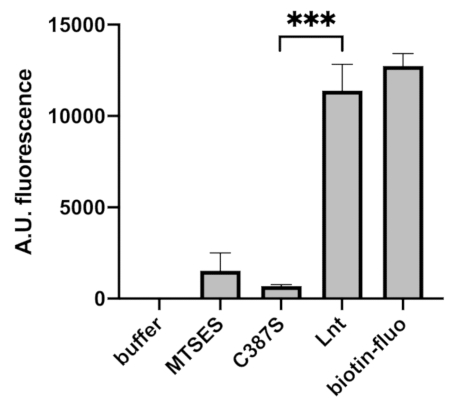
Figure 4: Fluorescence readout of the Lnt reaction in 384 well plate compatible with HTS. Enzymatic Lnt reactions were performed in 384 well plates at 37 °C. Biotin-fluorescein (biotin-fluo 0.19 µM) was used as control for fluorescence. Buffer was reaction buffer containing DMSO. Negative controls included inhibition with MTSES (10 mM) and an inactive variant of Lnt (C387S). Lnt enzyme was added at 0.5 ng/µL. Standard deviations were calculated for n = 3 experiments. *** P-value < 0.0005. Excitation at 485 nm, bandwidth 20 nm and emission at 535 nm, bandwidth 25 nm. Please click here to view a larger version of this figure.
