The miRNA pipeline, mirMachine, described above was applied to the test data for the fast evaluation of the performance of the pipeline. Only the high-confidence plant miRNAs deposited at miRBase v22.1 were screened against the chromosome 5A of IWGSC wheat RefSeq genome v224. mirMachine_find returned 312 hits for the nonredundant list of 189 high-confidence miRNAs with a maximum of 1 mismatch allowed (Table 1). mirMachine_fold classified 49 of them as putative miRNAs depending on the secondary structure evaluation. The highest represented group of miRNAs was miR9666 with a total of 18 miRNAs identified (Figure 1). Some miRNAs shared the same mature miRNA, but processed from a different pre-miRNA sequence. These miRNAs were renamed by the miRNA family name followed by a unique number, e.g., miR156-5p-1 and miR156-5p-2. Among the 49 putative miRNAs, 20 non-redundant mature miRNA sequences were identified. Some miRNAs can be transcribed from more than one locus resulting in a higher number of miRNAs represented. In the test data, miR9666-3p-5 was represented twice: one on the sense strand (at 602887137) and the other on the antisense strand (at 542053079). All locations are provided in the GitHub under the TestData output file named mature_high_conf_v22_1.fa.filtered.fasta.results.tbl. hairpins.tbl.out.tbl.
Expression evidence in one plant genome is sufficient, given the conservation of miRNAs in plants; however, a high-confidence miRNA dataset only provides a limited amount of data. Therefore, it is the user's preference to use the high-confidence and/or experimentally validated miRNAs as the reference dataset and skip the expression validation step, or to use all plant miRNAs available as the reference dataset and look for the expression evidence afterwards. Here, as the high-confidence miRNAs were used as the reference set, which had been validated experimentally in one of the plant genomes, the expression validation step was skipped for the test data.
mirMachine was benchmarked using monocot and dicot plants including Arabidopsis thaliana (Arabidopsis, TAIR10 release) and Triticum aestivum (wheat, IWGSC RefSeq v2). The performance of the homology-based and the sRNA-seq-based predictions was evaluated, and the results were compared with the miRDP225, an NGS-based miRNA prediction tool. Homology-based predictions were executed using the non-redundant list of plant mature miRNA sequences deposited at the miRbase v2226. sRNA-seq-based predictions were executed using the publicly available datasets; GSM2094927 for Arabidopsis and GSM1294661 for the wheat. In addition to raw results, the homology-based predictions were filtered for the expression evidence of mature miRNA and miRNA star sequences using the same sRNA-seq datasets.
Figure 2 shows the performance of each tool and the mirMachine settings on the two species. Sensitivity was calculated as the total number of known miRNAs identified divided by the total number of miRNAs identified. The results showed that mirMachine outperformed miRDP2 in terms of sensitivity and the true positive predictions in the Arabidopsis data. For the wheat data, mirMachine homology-based prediction, supported by expression evidence, provided better sensitivity than miRDP2. For both the genomes, miRDP2 predicted higher number of true positives compared to mirMachine sRNA-seq and homology-based predictions with expression evidence. It should be noted that miRDP2 lowers the expression threshold (RPM, reads per million) from 10 to 1 for the prediction of known miRNAs, resulting in higher true positive predictions. In general, the mirMachine can be used for the identification of both novel and known miRNAs. One advantage of the mirMachine is its ability to predict genome-wide distribution of the putative miRNAs without a limitation of specific tissues and conditions. Finally, the mirMachine is user-friendly and provides flexibility to adjust parameters such as number of hits, mismatches, length of miRNAs, and RPMs for specific research purposes. Taken together, the mirMachine provides accurate predictions for the putative miRNAs in the transcriptomes and the genomes of the plants.
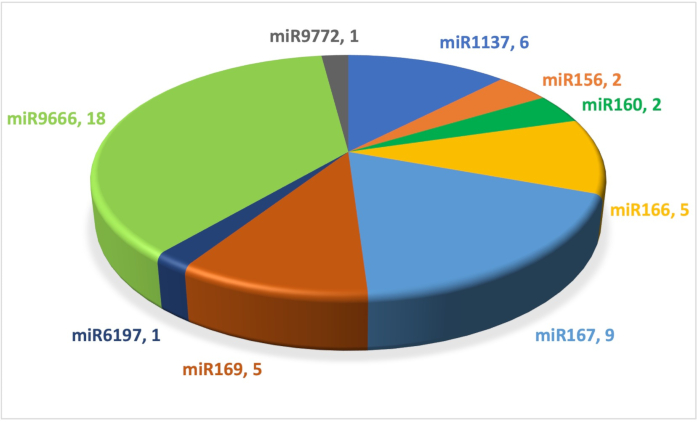
Figure 1: The distribution of miRNA families identified from the chromosome 5A of the IWGSC wheat reference genome v2. Data labels show the miRNA family and the number of miRNAs belonging to each miRNA family. Abbreviations: miRNA = microRNA; IWGSC = International Wheat Genome Sequencing Consortium. Please click here to view a larger version of this figure.
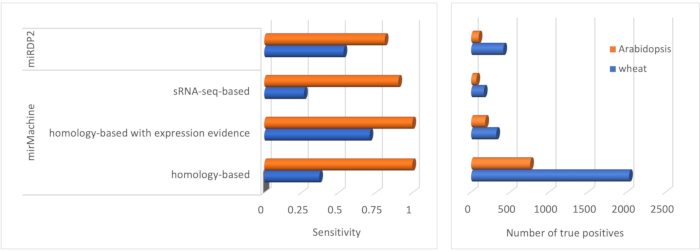
Figure 2: Performance assessment of the mirMachine. Comparisons of the sensitivity and the total number of known miRNAs predicted (true positives) are shown for the mirMachine with homology-based and sRNA-seq-based predictions and the miRDP2 software. Abbreviation: miRNA = microRNA. Please click here to view a larger version of this figure.
| Genome | Genome Size | Reference miRNA dataset | mirMachine_find hits | mirMAchine_fold hits | # of miRNA families |
| Test data | ~0.7 Gb | 189 | 312 | 49 | 9 |
| Chr5A |
Table 1: Statistics of the mirMachine. Test data are from the chromosome 5A of the IWGSC wheat reference genome v2. Abbreviations: miRNA = microRNA; IWGSC = International Wheat Genome Sequencing Consortium.
