The experiment's goal described below was to determine whether Bmp4 and Bmp7 form heterodimers (dimers composed of one Bmp4 ligand and one Bmp7 ligand), homodimers (composed of two Bmp4 or two Bmp7 ligands), or a mixture of each when they are co-expressed in X. laevis. Data shown in Figure 2 are extracted from a previously published study10. Figure 2A is a schematic showing cleavage products generated by proteolytic maturation of Bmp4 or Bmp7 homodimeric precursors or Bmp4/7 heterodimeric precursors. Bmp4 is cleaved at two sites within the prodomain to generate two prodomain fragments (dark green and yellow) and one dimeric ligand fragment (light green) that migrates at ~26 kDa on SDS-PAGE gels. Bmp7 is cleaved at a single site to generate one prodomain fragment (maroon) and one dimeric ligand fragment (pink) that migrates at ~35 kDa. Heterodimeric ligands migrate at ~30 kDa. The silver bar in the ligand dimers represents a myc-epitope tag inserted into this domain.
RNA encoding Bmp4myc or Bmp7myc precursors (200 pg), or both RNAs (100 pg of each) were injected into X. laevis embryos at the 2-4 cell stage. The embryos were cultured overnight at 16 °C until they reached the early gastrula stage. At this point, fluid was aspirated from the blastocele of 20 embryos in each group and sterile water was added to bring the volume to a total of 30 µL. Following deglycosylation, each sample was split into two tubes. Reducing sample buffer was added to one tube, and non-reducing sample buffer was added to the other. Samples were heated to 100 °C for 5 min. Proteins were then separated by SDS/PAGE and immunoblots were probed with antibodies that recognize the myc-epitope tag (Figure 2B). Under reducing conditions, a single band corresponding to cleaved Bmp4 monomers and a more slowly migrating band corresponding to cleaved BMP7 monomers were detected in lysates from embryos expressing only Bmp4 or Bmp7, respectively (Figure 2C, D, lower panel, lanes 1, 2). Both bands were detected in embryos co-expressing Bmp4 and Bmp7 (Figure 2C, D, lower panel, lane 3). Relatively equivalent amounts of Bmp4 and Bmp7 monomers were present in each of the three groups (lower panel, reducing). In this experiment, when proteins were separated under non-reducing conditions, a single mature Bmp4/7 heterodimer band of intermediate mobility was detected along with a trace amount of Bmp4 homodimer in embryos co-expressing Bmp4 and Bmp7 (Figure 2C, upper panel). From these results we conclude that if equivalent levels of Bmp4 and Bmp7 precursor proteins are expressed in Xenopus embryos, they preferentially form heterodimers, rather than either homodimer. In the experiment shown in Figure 2D, significantly more Bmp7 protein is present relative to Bmp4 (lower panel, reducing). As a result, in embryos expressing both Bmp7 and Bmp4 precursor proteins, Bmp4 homodimers are not detected and instead the available Bmp4 is present as a Bmp4/7 heterodimer, and the excess Bmp7 forms homodimers. While this experiment is not optimal, as the goal was to co-express the equivalent amount of Bmp4 and Bmp7 precursor, the results are still consistent with the conclusion that Bmp4 and Bmp7 preferentially form heterodimers over either homodimer when co-expressed.

Figure 1. Injection and aspiration needles. Photographs of representative needles that have been pulled but not clipped (A) pulled and clipped for use as an injection needle (B) or pulled and clipped for use as an aspiration needle (C) are shown. The arrow and arrowhead in (A) indicate the point at which the glass was clipped to generate a needle for injection and aspiration, respectively. Please click here to view a larger version of this figure.
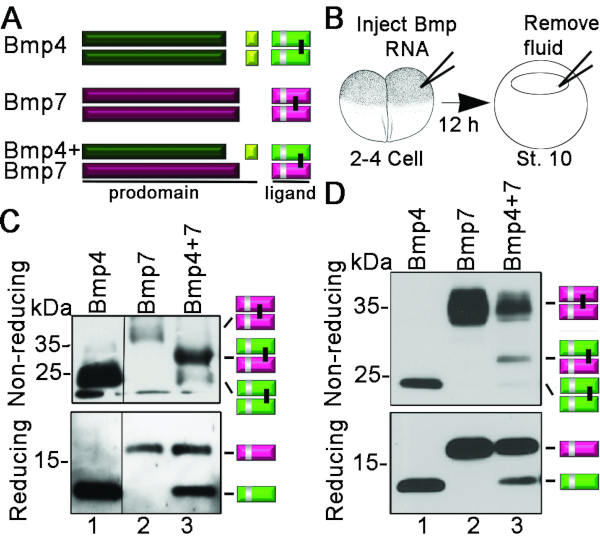
Figure 2. Analysis of cleaved Bmp ligands extracted from X. laevis blastocele fluid. (A) Cleavage products generated from Bmp4 and Bmp7 homodimeric or heterodimeric precursor proteins and position of myc epitope tag (silver bar) is shown schematically (B) Schematic diagram showing the timing of injection and blastocoele fluid extraction. (C-D) Xenopus embryos were injected with 200 pg of Bmp7 or Bmp4 RNA, or with Bmp4 and Bmp7 RNA mixed together (100 pg each). At the gastrula stage, fluid was extracted from the blastocoele of the same number of embryos in each experimental group. Proteins present in the blastocoele fluid were deglycosylated and separated by SDS-PAGE. Antibodies recognizing the myc-epitope tag were used to probe immunoblots. The relative position of immunoreactive ligand monomers and dimers is shown schematically to the right of each gel. Black line in (C) indicates removal of an intervening lane on the blot. Data shown are extracted from a previously published study10. Please click here to view a larger version of this figure.
| Solution | Composition |
| 0.2% Tricaine | 20 g of Tricaine dissolved in deionized water, adjust pH to 7.4 by addition of sodium bicarbonate |
| 10x MBS | 880 mM NaCl, 10 mM KCl, 25 mM NaHCO3, 100 mM HEPES (pH 7.5), 10 mM MgSO4, 0.14 mM Ca(NO3)2, 0.41 mM CaCl2; Adjust to pH 7.5 with NaOH. The 10x MBS stock can be stored at 4 °C and diluted as needed. |
| 2% cysteine solution | 2 g of cysteine per 100 mL of deionized water, adjust pH to 7.8-8.0 with sodium hydroxide. Make fresh each day. |
| 20x DeBoer's pond water | 100 mM NaCl, 1.3 mM KCl, 0.44 mM CaCl2; Adjust to pH 7.4 with NaHCO3. 20x stock of DeBoer’s pond water can be stored at 4 °C and diluted as needed. |
| 4x non-reducing sample buffer | 200 mM Tris pH 6.8, 8% SDS, 0.4% bromophenol blue, 40% glycerol. |
| 4x reducing sample buffer | 200 mM Tris pH 6.8, 8% SDS, 0.4% bromophenol blue, 40% glycerol, 14.4 M ß-mercaptoethanol |
| 5% Ficoll Solution | 25 g of Ficoll in 500 mL 0.1x MBS, adjust pH to 7.5 with sodium hydroxide. Store at 4 °C. |
| Embryo lysate buffer | 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 2.5 % NP-40. Store at 4 °C. |
| Human chorionic gonadotropin | Using a 3 mL syringe attached to a 19 G needle, inject 2.5 mL of sterile water through the rubber stopper of a vial of human chorionic gonadotropin (10,000 IU ). Store at 4 °C . |
| Testis buffer | 10% fetal bovine serum, 1% pen/strep (100U/mL penicillin, 100 mg/mL streptomycin in 1x MBS |
Table 1.
