1. Protein labeling
- Allow NHS-Rhodamine, 5-HT1A membrane fragments, and one 7 K MWCO spin desalting column to equilibrate at room temperature.
- Dissolve 1 mg of NHS-rhodamine in 100 µL of dimethyl sulfoxide (DMSO).
- Add 5 µL of 1 M sodium bicarbonate solution to increase the pH of 5-HT1AR solution to pH 8.
- Add 3.66 µL of the NHS-rhodamine solution to 50 µL of the 5-HT1AR solution and pipette gently up and down in a microcentrifuge tube.
NOTE: Ensure to have at least 10x molar excess of NHS-rhodamine. - Keep the mixture protected from light and put on rotator at room temperature for 1 h.
- Wash a 7 k MWCO spin column with 200 µL of 1x phosphate buffer saline (1x PBS) three times for 1.5 min at 1.5 RCF for each wash.
- Add the labeled protein to one column and balance the amount in another microcentrifuge tube.
- Spin down the labeled protein once for 5 min at 1.5 RCF.
- Take a UV-vis spectrum using a nanodrop spectrophotometer at 280 nm and 554 nm and calculate the labeling efficiency following the manufacturer's manual.
- Store the labeled protein covered at 5 °C until further use. The solution is stable for approximately a week after labeling.
2. GUVs with membrane-incorporated 5-HT 1A
- Preparation of materials and reagents
- Allow the protein, lipids and BSA (Bovine serum albumin) to equilibrate to room temperature.
- During this time, clean the coverslips by placing them in methanol and sonicating for 30 min at 40 °C. Ensure that the methanol completely covers the coverslips and the water level in the water bath is above the level of the methanol in the container.
NOTE: Methanol is toxic and should be handled in appropriate chemical hood. - Dry off the excess methanol on the coverslips with a gentle stream of air. Place the coverslip rack covered in a 40 °C oven for 15 min to ensure that the excess coverslips dry off.
- Begin the plasma cleaning process. First, place the coverslips into the plasma cleaner and close off the air intake valve to evacuate all the air inside the chamber.
- Once the chamber is under vacuum, clean the coverslips for 5 min using high RF power setting and a near complete vacuum, with only a slight air intake into the vacuum chamber. To ensure the proper level of plasma, adjust the opening of the vacuum chamber such that the resultant color of the plasma is a steady, bright pink.
NOTE: It is crucial when using air that the plasma remains a bright pink color for the duration of the plasma treatment step, as a darker purple color indicates that there is an improper amount of air in the chamber and will result in a suboptimal plasma treatment. - Once the 5 min have passed, shut off the RF power and release the vacuum.
NOTE: Upon removal from the plasma chamber, please ensure that the coverslips remain covered.
- Hydrogel preparation
- Combine 6 mg of ultra-low melting temperature agarose with 300 µL of ultrapure water (i.e., 2% (w/v) agarose).
NOTE: 2% agarose will be used to make protein-free GUVs. Agarose solution can be kept at 45 °C for two days. - Combine 9 mg of ultra-low temperature agarose with 300 µL of ultrapure water for 3 w/v% agarose by as prepared in step 3.1. 3% agarose will be used to make protein incorporated GUVs.
- Vortex the solution briefly before placing them on the 90 °C heat block for 10 min. Then, vortex the tube again before transferring it to a 45 °C heat block to keep it in the molten form until further use.
- Combine 6 mg of ultra-low melting temperature agarose with 300 µL of ultrapure water (i.e., 2% (w/v) agarose).
- Agarose and protein mixing
- Mix 21 µL of 3% agarose with the 7 µL of 5-HT1AR membrane fragments. Pipette up and down slowly many times to ensure adequate mixing. Then, incubate at 45 °C for 1 min.
- Hydrogel and lipid deposition
- For protein-free GUVs: Make a thin film on freshly plasma-cleaned coverslips using 20 µL of 2% agarose. Quickly drop another coverslip on top of the agarose droplet and gently slide the coverslips apart to make a thin film on both coverslips.
NOTE: This step is tricky in that the sliding of the droplet must occur while the agarose is still in the molten form. - For protein-incorporated GUVs: Pipette the protein/agarose mixture up and down one more time, and then deposit 20 µL of the 2% agarose on a plasma-cleaned coverslip. Follow the slip-casting directions as described above.
- Allow the agarose to gel protected from light for 30 min at room temperature.
- Deposit the lipids dropwise on top of the agarose layer. Use a total of 10 µL of 2 mg/mL of 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) with 0.4 Mol% 1,2-Dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) labeled with ATTO 488 (ATTO-488-DPPE) (or lipid mixture of interest) in chloroform on top of the agarose film. Deposit the droplets using a gas chromatography needle and spread one droplet at a time around via a gentle air stream.
NOTE: Caution is needed with this step to make a relatively uniform layer of lipids on top of the hydrogel. Also, chloroform is toxic and should be handled in appropriate chemical hood. - Assemble the Sykes-Moore (S-M) chambers by placing an O-ring on top of the coverslip, and then placing the top component of the chamber on top of the O-ring. Use the key provided by the manufacturer to assemble the chamber by screwing the chamber components together to seal the chamber and prevent any leakage.
NOTE: The top of the chamber should be tightened on the O-ring but caution is needed to ensure the coverslip stays intact as the coverslip can crack if the O-ring doesn't sit properly in the chamber. Also, ensure that the chamber is sealed tight enough such that the chamber does not leak when the swelling solution is added. Failure to tighten the chamber enough will result in leaks and loss of sample.
- For protein-free GUVs: Make a thin film on freshly plasma-cleaned coverslips using 20 µL of 2% agarose. Quickly drop another coverslip on top of the agarose droplet and gently slide the coverslips apart to make a thin film on both coverslips.
- Swelling and harvesting of vesicles
- Hydrate the entire system by gently pipetting 450 µL of 200 mM sucrose in 1x PBS and gently tapping the chambers to ensure adequate buffer coverage of the hydrogel-lipid layers.
NOTE: The sucrose solution can be replaced with a rehydration buffer containing biological probes of interest. - Place the chambers at 45 °C and cover the top part of the chamber with a coverslip to prevent evaporation. Allow the sample to swell, protected from debris and light for 1 h.
- Add 100 µL of 1 mg/mL BSA in ultrapure water into each well of a 96-well plate intended to be used. Incubate at room temperature for 1 h.
- Wash three times with ultra-pure water and once with 200 mM sucrose in 1x PBS.
- Finally, add 200 mM of glucose in 1x PBS until the addition of the GUV sample solution.
NOTE: BSA was used to block GUV adsorption. - After allowing the hydrogel to swell, gently shake and tap the chamber to dislodge any GUVs that may remain attached to the hydrogel surface. Then, carefully pipette up the GUV-sucrose solution.
NOTE: As an optional step to ensure all vesicles are detached from the surface, gently pipette some of the sucrose suspension back onto the hydrogel surface. - Move the suspension into a previously prepared microcentrifuge tube containing 700 µL of 200 mM glucose in 1x PBS.
NOTE: The density gradient will lead to settling of the vesicles to the bottom of the centrifuge tube. - Allow the vesicles to settle for another hour to ensure that the vesicles can sink to the bottom of the microcentrifuge tube, allowing for optimal collection.
- After the settling of GUVs in glucose, transfer 300 µL from the bottom of the centrifuge tube (the settled vesicles) into the previously prepared and BSA-treated 96-well plate to examine the vesicles under the confocal microscope.
NOTE: Be sure to avoid the very bottom of the microcentrifuge tube to minimize the amount of debris collected in the final sample.
- Hydrate the entire system by gently pipetting 450 µL of 200 mM sucrose in 1x PBS and gently tapping the chambers to ensure adequate buffer coverage of the hydrogel-lipid layers.
- Check the samples under the microscope.
- Shine a 488 nm laser on the sample (that allows us to visualize the membrane, as the bilayer has been labeled with ATTO-488-DPPE).
- Shine a 561 nm laser on the sample (that allows us to visualize the protein, since it has been labeled with NHS-Rhodamine).
NOTE: Caution is needed while imaging the sample as photooxidation can destabilize the vesicles. Vesicles were observed on the same day.
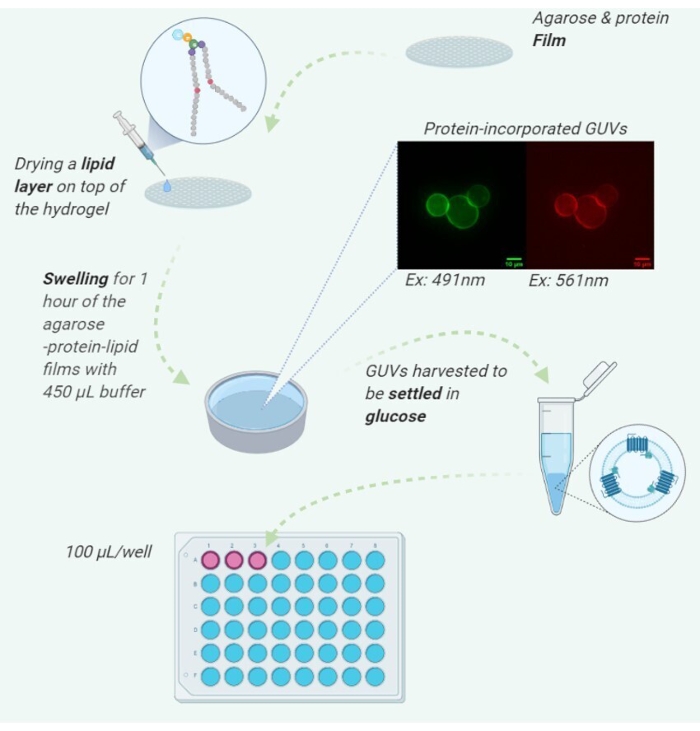
Figure 1: Illustration of the detailed protocol steps. Created with BioRender.com Please click here to view a larger version of this figure.
The concentration of protein was measured, and the degree of labeling was calculated as the molar ratio between the dye and the protein to be 1:1. By examining the GUVs using confocal microscopy, we were able to confirm successful formation and protein integration of the vesicles. The lipids were labeled with 0.4 mol% ATTO 488-DPPE, and the protein was covalently labeled via rhodamine NHS-ester modification of primary amines. Figure 2a and Figure 2b show a protein-incorporated vesicle in the ATTO 488 and rhodamine channels, respectively. All micrographs have been dark current and flatfield corrected. Figure 2c and Figure 2d show a negative control GUV with no protein incorporated. Figure 3a and Figure 3b show a protein incorporated GUV with line intensity profiles given by the dashed white line of the same vesicle in both channels. The line intensity profile shows a two-dimensional plot of the intensities of the pixels along the white drawn line within the image. The x-axis is the distance along the line and the y-axis is the pixel intensity. ImageJ software was used to plot the profile intensity of the indicated line.
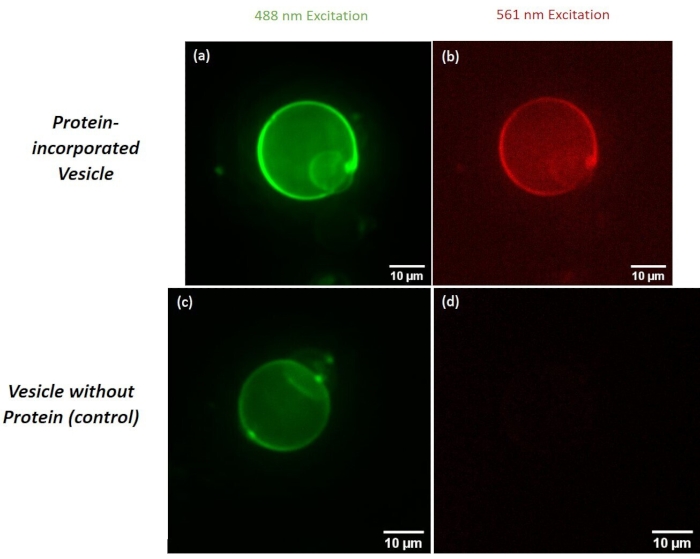
Figure 2: Micrographs comparing protein incorporated GUVs and GUVs without protein (control). Micrographs (a) and (b) show protein incorporated GUV fluorescence with the respective ATTO 488 and rhodamine channels, respectively. Micrographs (c) and (d) show a protein omitted GUV when excited with ATTO 488 and rhodamine channels, respectively. Please click here to view a larger version of this figure.
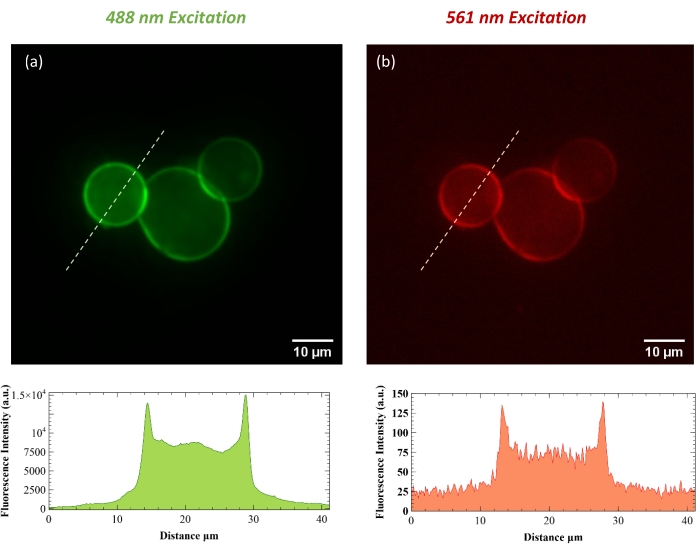
Figure 3: Top row shows micrographs of protein incorporated GUVs in ATTO 488 (a) and rhodamine (b) channels. Line intensity profiles for the indicated white-dashed lines are below. The analysis was performed using ImageJ software. Please click here to view a larger version of this figure.
