1. Media and buffer preparation
- Prepare KB and LB media and agar plates as given in Table 1, and autoclave at 121 °C, 15 psi, 20 min.
- Add 50 µg/mL filter-sterilized kanamycin and 300 µM filter-sterilized 2,6-diaminopimelic acid (DAP) to the autoclaved LB medium before culturing E.coli WM3064 + pRL27.
- Add 50 µg/mL filter-sterilized kanamycin to the autoclaved KB agar to pour plates needed for selecting successful B. gladioli Lv-StA transconjugants.
- Prepare 1x phosphate-buffered saline (PBS) by mixing the following components: NaCl 8 g/L, KCl 0.201 g/L, Na2HPO4 1.42 g/L, and KH2PO4 0.272 g/L. Dissolve the salts in distilled water and autoclave the mixture at 121 °C, 15 psi, 20 min before use. Store at room temperature.
- Prepare a 2x Bind-and-wash buffer by dissolving the following components: 10 mM Tris-HCl (pH 7.5), 1 mM ethylenediamine tetraacetic acid (EDTA), and 2 M NaCl in distilled water. Filter-sterilize the mixture before use. Store at room temperature.
- Prepare 1x Low-TE by dissolving 10 mM Tris-HCl (pH 8.0) and 0.1 mM EDTA in double-distilled water. Sterilize by autoclaving at 121 °C, 15 psi, 20 min. Store at room temperature.
2. Conjugation to generate the transposon mutant library
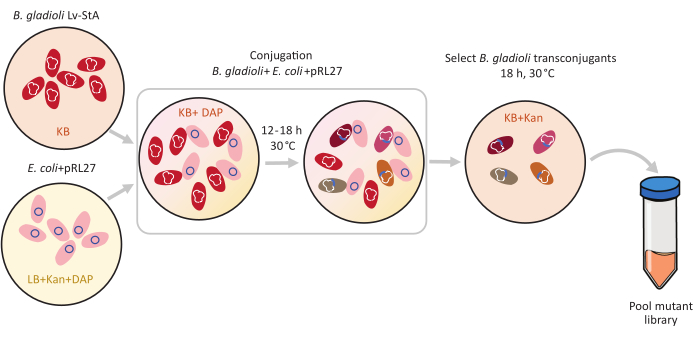
Figure 1: Conjugation protocol steps. The conjugation recipient Burkholderia gladioli Lv-StA (red) and donor Escherichia coli containing the pRL27 plasmid (pink) are grown in KB agar and LB, respectively, supplemented with kanamycin and DAP. After conjugative transfer of the plasmid for 12-18 h at 30 °C, the transconjugant B. gladioli cells are selected on KB containing kanamycin and pooled together. Abbreviations: DAP = 2,6-diaminopimelic acid; Kan = kanamycin. Please click here to view a larger version of this figure.
- Under a sterile hood, inoculate a fresh donor culture of Escherichia coli WM3064 + pRL27 in 10 mL of LB medium supplemented with kanamycin and DAP. Inoculate Burkholderia gladioli Lv-StA recipient cells in 5 mL of KB medium. Incubate the cultures at 30 °C overnight on a shaker at 250 rpm.
- After overnight growth, centrifuge 4 mL of each of the cultures at 9,600 × g for 6 min to pellet the cells. Discard the supernatant.
- Under a sterile hood, wash the pelleted cell cultures in KB medium containing DAP and finally resuspend the cultures separately in 4 mL of KB + DAP medium.
- In a fresh 15 mL tube, mix 250 µL of the washed E. coli donor cells with 1 mL of the washed B. gladioli Lv-StA recipient cells.
- Spot 10 µL of this conjugation cell mixture on KB agar plates containing DAP. Allow the plate to rest undisturbed in the sterile hood at room temperature for 1 h. Then, incubate the plates with the conjugation spots at 30 °C for 12-18 h.
NOTE: The conjugation period can be adjusted according to the target species. However, a long conjugation period increases the risk for double insertions or plasmid integration into the genome. For slow-growing bacteria, allow for longer conjugation periods. - After incubation, add 2-4 mL of 1x PBS into the plates under a sterile hood and use a cell scraper to release the grown bacterial conjugation spots from the agar. Pipette the conjugated-cell-mix into 2 mL microfuge tubes.
- Pellet the cells by centrifuging at 9,600 × g for 2 min. Discard the supernatant and wash the pellet twice in 1 mL of 1x PBS by pipetting up and down. Resuspend the final pellet in 1200 µL of 1x PBS. Make dilutions before plating if the number of cells in the mixture is above 1 × 104.
- Mix well and spread 200 µL of the cell mixture on large KB agar plates (6 or more, if required) supplemented with kanamycin and incubate at 30 °C overnight.
NOTE: Target mutant colonies appear within 30 h on the selective agar plates. Due to the antibiotic resistance marker, only mutated colonies appear on the selective agar plate. Therefore, all colonies are expected to be successful transconjugants. - Count the total number of transconjugant colonies on three plates and extrapolate to calculate the approximate number of mutants obtained in all the plates. To increase the chances of obtaining a representative library, ensure that the total number of colonies is several fold higher than the total number of genes in the genome. To confirm the success of conjugation, perform a PCR targeting the insertion cassette using 10-20 sample colonies, as described in section 3.
NOTE: The aim is to ensure that the number of colonies is at least 10-fold the number of genes in the whole genome, in this case, >75,000 mutants. However, it is generally challenging to accurately estimate the number of colonies that would correspond to a fully representative library. The number of unique genes mutated is not evident at this point, given that disruptions in essential genes are not captured, there are often multiple different mutation sites for the same gene, and mutations generated with Tn5 transposons are not entirely random. - Under a sterile hood, scrape colonies from the plates by adding 1-2 mL of 1x PBS on the agar. Pool the cell mixture scraped off from the plates into 50 mL tubes. Vortex the library to mix thoroughly and then split 4 mL of the pooled mutant library into several cryotubes. Add 1 mL of 70% glycerol to the tubes and store at -80 °C.
3. PCR and gel electrophoresis to confirm successful insertions in B. gladioli Lv-StA
- To confirm the presence of the insertion, pick individual mutant colonies from the selection plates in step 2.9 and perform a PCR targeting the insertion cassette using the primers listed in Table 2. Prepare the PCR master mix according to Table 3 and set conditions in the thermal cycler as described in Table 4.
- Run the PCR products on a 1.6% agarose gel by electrophoresis (250 V, 40 min) to check if the amplified DNA fragments are of the expected length of 1580 bp.
4. Mutant pool infection on beetle eggs
- Library washing steps
- Thaw an aliquot of the prepared mutant library on ice. Centrifuge at 2,683 × g for 10 min and remove the supernatant. Under a sterile hood, wash the cells with 4 mL of 1x PBS to remove any remaining medium from the cells. Resuspend the cells in 4 mL of 1x PBS.
- Count the number of cells in an aliquot of the library using a cell counting chamber. Dilute a part of the library to 2 × 106 cells/µL in 1x PBS.
- Vortex the library aliquot thoroughly to mix the whole library homogenously before taking the required volume.
- Egg clutch sterilization and in vivo infection
- Select an L. villosa egg clutch. Count the number of eggs and continue if the clutch contains more than 100 eggs.
- Sterilize the entire egg clutch.
- Add 200 µL of 70% ethanol and gently wash the eggs for 5 min. Remove the ethanol and wash the eggs twice with autoclaved water.
- Add 200 µL of 12% bleach (NaOCl) and gently wash the eggs for 30 s. Remove the bleach immediately and wash the eggs again three times with 200 µL of autoclaved water.
- Infect 2 × 106 cells/µL of the washed mutant library on the sterilized egg clutch (2.5 µL per egg).
- Two days after the infected beetle larvae hatch, collect 100 2nd instar larvae per 1.5 mL microfuge tube and store at -80 °C.
- In vitro mutant library control
- Under a sterile hood, inoculate 250 µL of 2 × 106 cells/µL of the washed mutant library in 10 mL of KB medium containing kanamycin.
- Incubate the in vitro mutant culture at 30 °C for 20 h.
NOTE: Calculate the duration of incubation to match the approximate number of generations of WT B. gladioli Lv-StA in vivo during colonization. - After the 20 h incubation, add an equal volume of 70% glycerol to the in vitro mutant culture and store it at -80 °C.
5. Infected beetles and in vitro mutant library DNA extraction
NOTE: DNA extractions were performed using a DNA and RNA purification kit according to the manufacturer's protocol briefly outlined below.
- Homogenize pooled larvae (maximum of 4 mg per microfuge tube) by adding 1-2 mL of liquid nitrogen and crushing with a pestle.
- Thaw the in vitro grown mutant cultures from glycerol stocks on ice. Pellet the cells by centrifuging at 9,600 × g for 10 min before cell lysis.
- Add 300 µL of Tissue and Cell lysis solution to the in vitro and in vivo samples. Add 5 µL of 10 mg/mL Proteinase K, incubate the mix at 60 °C for 15 min, and then place on ice for 3-5 min.
- Add 150 µL of protein precipitation reagent to the lysates and vortex thoroughly. Pellet the protein debris by centrifuging at 9,600 × g for 10 min.
- Transfer the supernatant to a 1.5 mL microfuge tube. Add 500 µL of isopropanol to the supernatant and gently invert the tubes at least 40 times before incubating at -20 °C for 1 h or overnight.
- Pellet the precipitated DNA by centrifuging at 9,600 × g for 10 min. Discard the supernatant and add ice-cold 70% ethanol to the DNA pellet.
- Centrifuge at ≥10,000 × g for 5 min. Discard the supernatant and leave the samples to air-dry for at least 1 h.
- Resuspend the DNA from the in vitro and in vivo samples in 100 µL of Low-TE buffer.
- Store the samples at -20 °C.
6. Sequencing library preparation
NOTE: The protocol and reagents for DNA library preparation are adapted and modified from the instructions provided by the manufacturer of the DNA library preparation kit.
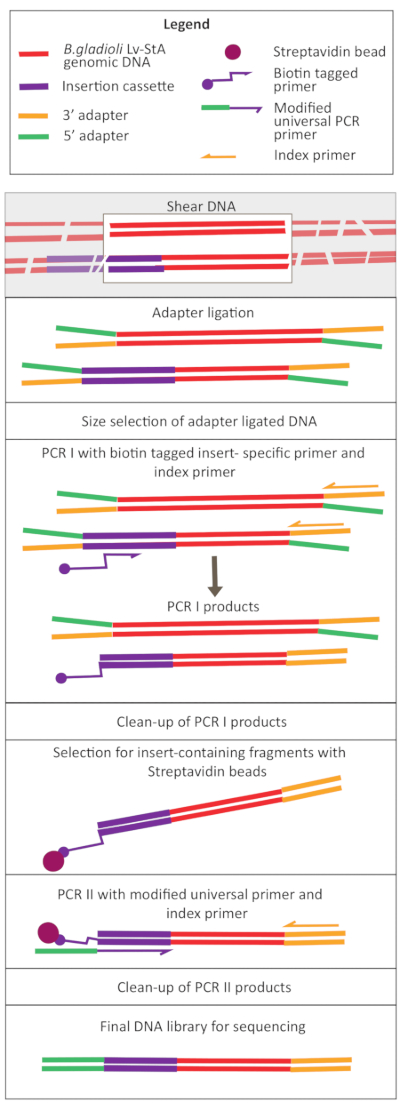
Figure 2: Schematic of the DNA library preparation steps. After shearing and adapter ligation, the modified protocol includes a streptavidin bead-selection step to enrich DNA fragments containing the insertion cassette. Please click here to view a larger version of this figure.
- Dilute the samples to 20 ng/µL concentration and volume of 100 µL and keep them on ice.
- Shear in vivo and in vitro sample DNA using an ultrasonicator. Set the ultrasonicator at 70% power. Vortex the samples briefly and shear for 1 min 30 s.
NOTE: The settings for the ultrasonicator will differ among instruments. In this case, the fragment size was 200-400 bp, which is appropriate for this sequencing approach of 150 bp, paired-end (see step. 9.1). The shearing parameters can be adjusted according to the experimenter's requirements. - Check if the DNA was sheared to the desired size range (in this case, 200-400 bp). Load 5 µL of the unsheared and sheared DNA after mixing with gel loading dye in a 1:1 ratio on a 1.6% agarose gel run at 250 V for 40 min (Figure 3A,B).
- Preparation of fragment ends required for adapter ligation
- To 50 µL of the sheared DNA, add the end preparation reagents given in the library preparation kit: 3 µL of the enzyme mix and 7 µL of reaction buffer and mix well by pipetting. Set a thermal cycler with a heated lid at ≥ 75 °C and incubate the samples for 30 min at 20 °C and 30 min at 65 °C. Hold at 4 °C.
- Adapter ligation
- For adapter ligation, add the following reagents to the products of the end preparation step: 30 µL Ligation Master Mix, 1 µL Ligation Enhancer, and 2.5 µL diluted Adapter. Mix thoroughly by pipetting and incubate the sample for 15 min at 20 °C in the thermal cycler with the heated lid off.
- After 15 min, add 3 µL of the enzyme (uracil DNA glycosylase + DNA glycosylase-lyase Endonuclease VIII) (see the Table of Materials). Mix well by pipetting and incubate the sample for 15 min at 37 °C in a thermal cycler with the lid heated at ≥47 °C.
NOTE: The protocol can be paused at this step, and the samples can be stored at -20 °C.
- Size selection of adapter-ligated DNA targeting fragments of 250 bp
- Vortex the magnetic bead solution (see the Table of Materials) and place it at room temperature for 30 min before use.
- Add 0.3x of beads to 96.5 µL of the ligated DNA mixture and mix by pipetting thoroughly. Incubate the bead mixture for 5 min.
NOTE: The presence of salts and polyethylene glycol in the bead mixture facilitates the precipitation of DNA fragments on the beads. A low ratio of beads to DNA molecules leads to the binding of only larger DNA fragments to the beads. In this case, DNA fragments above 250 bp in length are bound to the beads. - Place the tubes on a magnetic stand to pull down the beads and remove DNA fragments of unwanted size. Let the beads settle for 5 min and then transfer the clear supernatant to a new microfuge tube (keep the supernatant).
- Add 0.15x of fresh beads to the supernatant and mix by pipetting well. Incubate the bead mixture for 5 min and then place the tubes on a magnetic stand to pull down the beads bound to the target DNA. Wait for 5 min and then discard the supernatant (keep the beads).
NOTE: This ratio of beads to DNA leads to the binding of fragments of the desired 250 bp size. - With the beads on the magnetic stand, add 200 µL of 80% ethanol (freshly prepared) and wait for 30 s. Pipette out and discard the ethanol wash carefully without disturbing the beads on the magnetic stand. Repeat this step.
- After the last wash, remove traces of ethanol from the beads and then air-dry the beads for 2 min until they appear glossy but not completely dried out. Do not over-dry the beads.
- Remove the tubes from the magnetic stand and add 17 µL of 10 mM Tris-HCl or 0.1x TE (Low-TE). Mix by pipetting ~10 times and incubate the mixture at room temperature for 2 min.
- Place the tubes back on the magnetic stand and wait for 5 min. Once the beads have settled down, transfer the DNA supernatant to a new tube.
- PCR I to add biotin tag to DNA fragments containing the insertion cassette
- Add a biotinylated primer tag to the DNA fragments containing the Tn5-insertion cassette by using the transposon-specific biotinylated primer (Table 5) and an index primer. Prepare the PCR master mix according to Table 6 and follow the PCR conditions for the thermal cycler listed in Table 7.
- Clean-up of PCR I without size selection
- Vortex 0.9x beads and place them at room temperature for at least 30 min before clean-up.
- Add 0.9x beads to the PCR products and mix thoroughly.
- Place the beads on a magnetic stand to pull down the beads.
- Remove the clear supernatant and wash the bead-bound-DNA with 200 µL of freshly prepared 80% ethanol twice.
- Remove the ethanol after the wash steps and air dry the beads until they look glossy but not too dry.
- Add 32 µL of 10 mM Tris-HCl or 0.1X TE (Low-TE) and incubate the beads for 5 min. Place the mixture back on the magnetic stand and transfer the supernatant to a fresh microfuge tube.
- Binding biotinylated DNA fragments to streptavidin beads
- Resuspend 32 µL of streptavidin beads in 1x Bind-and-wash buffer. Wash the beads with the buffer three times while placed on a magnetic stand.
- Add 32 µL of 2x Bind-and-wash buffer and resuspend the beads. To this, add 32 µL of the cleaned-up PCR 1 products. Mix thoroughly and incubate at room temperature for 30 min.
- Place the bead-DNA mixture on a magnetic stand for 2 min. Pipette out the supernatant as biotin-tagged DNA containing the insertion edge binds to streptavidin on the beads.
- Wash the beads with 500 µL of 1x Bind-and-wash buffer and then wash the beads with 200 µL of Low-TE. Resuspend the DNA-bound beads in 17 µL of Low-TE.
- PCR II to add adapters to the fragments containing the insertion cassette edge
- Prepare a master mix, as shown in Table 8, using the index primers and modified universal PCR primers listed in Table 5. Add 15 µL of the DNA-bound streptavidin beads from the previous step to the PCR mix. See Table 7 for the thermal cycler conditions.
- Clean up the PCR products without size selection as given in step 6.8 of this protocol. Elute the final DNA products in 30 µL of molecular-grade water.
- Store the samples at -20 °C and use them for sequencing.
7. Sequencing and analysis
- Sequence the library using high-throughput sequencing technology. Adjust the sequencing depth depending on the transposon library size, as noted below. Assess the read quality with FastQC7. Select reads containing the Tn5-insertion edge on the 5' end of the read and remove the insertion edge sequence using Cutadapt8 and/or Trimmomatic9.
NOTE: Here, a paired-end sequencing approach was used to target 150 bp per read and a total of 8 Mio reads. To obtain a representative dataset, ensure that the total number of sequenced reads exceeds the maximum possible number of mutants in the library, i.e., the total estimated number of colonies from step 2.9. As a reference, this protocol aimed for 40-fold of the maximum possible library size. Other successful studies using Tn-seq for a similar purpose sequenced a total number of reads close to 25-fold of the actual number of unique insertions in the corresponding mutant library22,23. - Considering that mutations at the ends of genes are not functionally disruptive, trim 5% off both ends of gene annotations of the reference genome GFF file. Map the trimmed reads to the reference genome using Bowtie210.
- Calculate the number of insertions from the number of unique 5' positions in the alignment BAM file.
- Using FeatureCounts11, obtain the number of hit genes for each replicate sample.
- Using the DESeq212 package in RStudio, calculate the difference in mutant abundances between different conditions.
Host-associated bacteria can employ several factors to establish an association, including those mediating adhesion, motility, chemotaxis, stress responses, or specific transporters. While factors important for pathogen-host interactions have been reported for several bacteria13,14,15,16,17,18, including members of the genus Burkholderia19,20, fewer studies have explored the molecular mechanisms used by beneficial symbionts for colonization21,22,23. Using transposon insertion sequencing, the aim was to identify molecular factors that enable B. gladioli to colonize L. villosa beetles.
Transposon-mediated mutagenesis was performed using the pRL27 plasmid, which carries a Tn5 transposon and a kanamycin resistance cassette flanked by invert repeat sites. The plasmid was introduced into the target B. gladioli Lv-StA cells by conjugation with the plasmid donor E. coli WM3064 strain (as shown in Figure 1). After conjugation, the conjugation mix containing B. gladioli recipient and E. coli donor cells were plated on selective agar plates containing kanamycin. The absence of DAP on the plates eliminated the donor E. coli cells, and the presence of kanamycin selected for successful B. gladioli Lv-StA transconjugants. The pooled B. gladioli Lv-StA mutant library obtained from harvesting the 100,000 transconjugant colonies was prepared for sequencing using a modified DNA library preparation kit and custom primers. Figure 2 highlights the DNA library preparation steps. Sequencing yielded 4 Mio paired reads; 3,736 genes out of 7,468 genes in B. gladioli Lv-StA were disrupted.
To identify mutants that were colonization-defective in the host, the B. gladioli Lv-StA mutant library was infected on the beetle eggs and grown in vitro in KB medium as a control. The in vivo colonization bottleneck size was calculated before the experiment. A known number of B. gladioli Lv-StA cells was infected on beetle eggs, and the number of colonizing cells in freshly hatched first instar larvae was obtained by plating a suspension from each larva and counting colony-forming units per individual. These calculations were done to ensure that the number of colonizing cells is enough to assess all or a high percentage of the mutants in the library for their ability to colonize the host. Additionally, the growth time between in vitro and in vivo conditions was normalized based on the number of bacterial generations to make these samples comparable.
After the eggs hatched, 1,296 larvae were collected in 13 pools. The corresponding in vitro mutant cultures were grown and stored as glycerol stocks. DNA of the in vivo and in vitro grown mutant libraries was extracted and fragmented in an ultrasonicator. Figure 3 shows the size distribution of the sheared DNA, where the majority of the fragments span between 100 and 400 bp, as expected. This step was followed by the modified DNA library preparation protocol for sequencing. At each step of the protocol, the concentration of remaining DNA was checked to ensure that the steps were performed correctly and to track losses of DNA. A quality check (see the Table of Materials) before sequencing revealed that the DNA libraries contained unexpectedly large (>800 bp) DNA fragments, and this was more pronounced in the in vivo libraries. Given the difficulty in optimizing the clustering of fragments in the sequencing lanes, it was necessary to increase the sequencing depth to 10 Mio paired reads in the in vivo libraries to attain the desired number of reads. The analysis of the sequencing results revealed that an average of 4 Mio reads in the in vivo libraries and 3.1 Mio reads in the in vitro libraries contained the Transposon edge in the 5' end of Read-1 (Table 9), which was satisfactory for this experiment. The distribution of the 24,224 unique insertions across the B. gladioli genome in the original library is shown in Figure 4. An analysis carried out using DESeq2 revealed that the abundances of 271 mutants were significantly different between the in vivo and in vitro conditions.
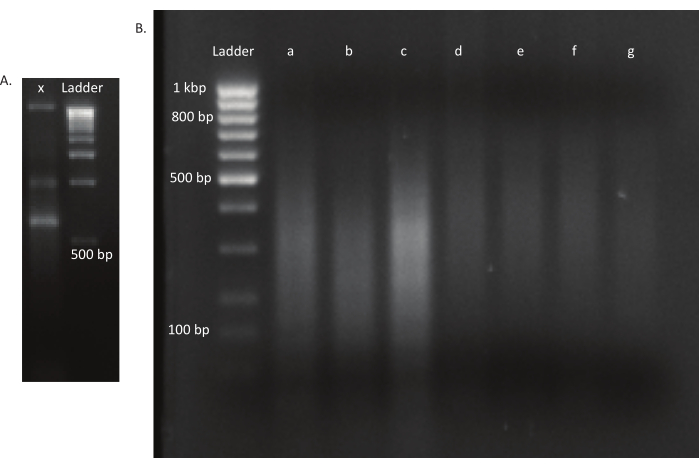
Figure 3: Agarose gels of a mutant and DNA libraries. (A) Agarose gel with unsheared DNA of a mutant in lane x and a 1 kbp ladder for scale. (B) Gel with sheared DNA library. The band sizes of the ladder in the first lane are indicated on the left side. The first three lanes a, b, and c contain sheared DNA fragments of the in vivo libraries. Lanes d, e, f, and g contain sheared DNA fragments of the in vitro libraries. Please click here to view a larger version of this figure.
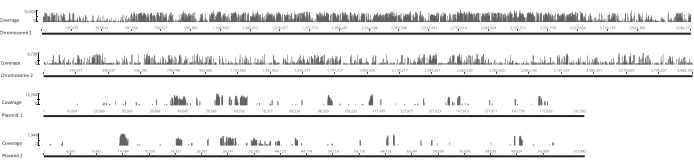
Figure 4: Location of unique insertion sites in the original library across the four replicons in the Burkholderia gladioli Lv-StA genome. Each bar along the x-axis is located at a site of insertion. The height of a bar along the y-axis corresponds to the number of reads associated with that site. Note that the two chromosomes and two plasmids are shown in full length and thus have different scales on the x-axis. Please click here to view a larger version of this figure.
| King’s B medium/ agar | |
| Peptone (soybean) | 20 g/L |
| K2HPO4 | 1.5 g/L |
| MgSO4.7H2O | 1.5 g/L |
| Agar | 15 g/L |
| Dissolved in distilled water | |
| LB medium/agar | |
| Tryptone | 10 g/L |
| Yeast extract | 5 g/L |
| NaCl | 10 g/L |
| Dissolved in distilled water | |
Table 1: Media components.
| No. | Primers | Sequence | PCR annealing temp. (°C) | |
| 1 | tpnRL17–1RC | 5’-CGTTACATCCCTGGCTTGTT-3’ | 58.2 | |
| 2 | tpnRL13–2RC | 5’-TCGTGAAGAAGGTGTTGCTG-3’ | ||
Table 2: Primers to confirm the success of conjugation.
| Component | Volume (μL) |
| HPLC-purified water | 4.92 |
| 10x Buffer S (high specificity) | 1 |
| MgCl2 (25 mM) | 0.2 |
| dNTPs (2 mM) | 1.2 |
| Primer 1 (10 pmol/µL) | 0.8 |
| Primer 2 (10 pmol/µL) | 0.8 |
| Taq (5 U/µL) | 0.08 |
| Mastermix total | 9 |
| Template | 1 |
Table 3: PCR master mix to confirm the success of conjugation. Abbreviations: HPLC = high-performance liquid chromatography; dNTPs = deoxynucleoside triphosphate.
| Steps | Temperature °C | Time | Cycles |
| Initial Denaturation | 95 | 3 min | 1 |
| Denaturation | 95 | 40 s | |
| Annealing | 58.2 | 40 s | 30 to 35 |
| Extension | 72 | 1-2 min | |
| Final Extension | 72 | 4 min | 1 |
| Hold | 4 | ∞ | |
Table 4: PCR conditions to confirm the success of conjugation.
| Primers | Sequence | Tm °C | Use | Source | |
| Transposon-specific biotinylated primer | 5’-Biotin-ACAGGAACACTTAACGGCTGACATG -3’ |
63.5 | 6.7.1. PCR I | Custom | |
| Modified Universal PCR primer | 5’- AATGATACGGCGACCACCGAGATC TACACTCTTTCCCTACACGACGCTC TTCCGATCTGAATTCATCGATGAT GGTTGAGATGTGT – 3’ |
62 | 6.10.1. PCR II | Custom | |
| Index primer | Refer to the manufacturer’s manual | 6.7.1. PCR I & 6.10.1. PCR II | NEBNext Multiplex Oligos for Illumina (Index primers set 1) | ||
| Adapter | Refer to the manufacturer’s manual | 6.5. Adapter ligation | NEBNext Ultra II DNA library prep kit for Illumina | ||
Table 5: Primers and adapter for PCR I and II during DNA library preparation.
| PCR mix | (µL) |
| Adapter-ligated DNA fragments | 15 |
| NEBNext Ultra II Q5 master mix | 25 |
| Index primer (10 pmol/ µL) | 5 |
| Transposon specific biotinylated primer (10 pmol/ µL) | 5 |
| Total volume | 50 |
Table 6: DNA library preparation-PCR I master mix.
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | 6 to 12 |
| Annealing | 65 °C | 30 s | |
| Extension | 72 °C | 30 s | |
| Final Extension | 72 °C | 2 min | 1 |
| Hold | 16 °C | ∞ | |
Table 7: DNA library preparation-PCR I and II conditions.
| PCR mix | (µL) |
| Bead-selected DNA | 15 |
| NEBNext Ultra II Q5 master mix | 25 |
| Index primer | 5 |
| Modified universal PCR primer | 5 |
| Total volume | 50 |
Table 8: DNA library preparation-PCR II master mix.
| Libraries | Invivo-1 | Invivo-2 | Invivo-3 | Invitro-1 | Invitro-2 | Invitro-3 | Original library | |
| No. of reads (PE) | 56,57,710 | 39,19,051 | 30,65,849 | 35,73,494 | 28,83,440 | 36,61,956 | 46,09,410 | |
| No. of reads containing Tn – edge on 5’ end of Read-1 | 54,15,880 | 37,31,169 | 29,36,247 | 33,00,499 | 27,35,705 | 33,50,402 | 41,53,270 | |
| Bowtie2 overall alignment rate (%) (Read-1 only) | 95.53% | 83.71% | 89.87% | 80.79% | 78.00% | 73.06% | 74.92% | |
| Number of unique insertions | 8,539 | 4,134 | 7,183 | 18,930 | 18,421 | 20,438 | 24,224 | |
| Number of genes hit | 1575 | 993 | 1450 | 2793 | 2597 | 3037 | 3736 | |
Table 9: Summary of sequencing output and transposon insertion frequency per library. Abbreviation: PE = paired-end.
