The example ribosome profiling datasets were deposited in the GEO database under the accession number GSE131074. All the files and codes used in this protocol are available from Supplemental files 1–4. By applying RiboCode to a set of published ribosome profiling datasets23, we identified the novel ORFs actively translated in MCF-10A cells treated with control and EIF3E siRNAs. To select the RPF reads that are most likely bound by the translating ribosomes, the lengths of the sequencing reads were examined, and a metagene analysis was performed using the RPFs that mapped on the known translation genes. The frequency distribution of the lengths of the reads showed that most RPFs were 25-35 nt (Figure 1A), corresponding to a nucleotide sequence covered by the ribosomes as expected. The P-site locations for different lengths of RPFs were determined by examining the distances from their 5' ends to the annotated start and stop codons, respectively (Figure 1B). The RPF reads within 28-32nt displayed strong 3-nt periodicity, and their P-sites were at the +12th nt (Supplemental file 1).
RiboCode searches for the candidate ORFs from a canonical start codon (AUG) or alternative start codons (optional, e.g., CUG and GUG) to the next stop codon. Then, based on the mapping results of RPFs within the defined range, RiboCode assesses the 3-nt periodicity by evaluating whether the number of in-frame RPFs (i.e., their P-sites allocated on the first nucleotide of each codon) is greater than the number of out-of-frame RPFs (i.e., their P-sites allocated on the second or third nucleotide of each codon). We identified 13,120 genes potentially translating ORFs with p < 0.05, among them 10,394 genes (70.8%) encoding annotated ORFs, 168 (1.1%) genes encoding dORFs, 509 (3.5%) genes encoding uORFs, 939 (6.4%) genes encoding upstream or downstream ORFs overlapped with known annotated ORFs (Overlapped), and 68 (0.5%) protein-coding genes encoding novel ORFs, and 2,601 (17.7%) previously assigned as noncoding genes encoding novel ORFs (Figure 2 and Supplemental file 3)
Comparing sizes of different ORFs showed that uORFs and overlapped ORFs are shorter (195 and 188 nt on average, respectively) than annotated ORFs (~1,771 nt). The same trend was also observed for novel ORFs (670 and 385 nt on average for novel PCGs and novel nonPCGS, respectively) and dORFs (~671 nt) (Figure 3). Together, those noncanonical ORFs (unannotated) identified by RiboCode tended to encode peptides that are smaller than those known annotated ORFs.
Relative RPF counts were calculated for each ORF to assess the function of EIF3 in the processes of translation. The results suggested that the ribosome densities of uORFs were significantly higher in EIF3E-deficient cells than in control cells (Figure 4). As many uORFs were reported to exert inhibitory effects on the translation of downstream coding ORFs, we further examined whether the EIF3E knockdown alters the global densities of RPFs downstream of the start codons (Figure 5). The metagene analysis, in which many ORFs' profiles were aligned and then averaged, revealed that a mass of ribosomes stalled between codons 25 and 75 downstream of the start codon, suggesting that the translation elongation might be blocked early in EIF3E-deficient cells. Further investigations are warranted to examine whether the signal-to-noise ratio or the changes in translation efficiency of ORFs contribute to the increase in uORF RPKM and the accumulation of ribosomes between codons 25 to 75 in the absence of EIF3E, that is, whether the 1) less contamination (or good library quality) or 2) active translation (or ribosome pausing) in the samples without EIF3E results in more reads in uORFs and in the defined region between the 25th and 75th codons.
Finally, RiboCode also provides visualization for densities of the P-sites of RPFs on desired ORF, which could help users to examine the 3-nt periodicity patterns and densities of RPFs. For example, Figure 6 presents the RPF densities on an uORF of PSMA6 and a dORF of SENP3-EIF4A1; both were validated by published proteomics data23 (data not shown).
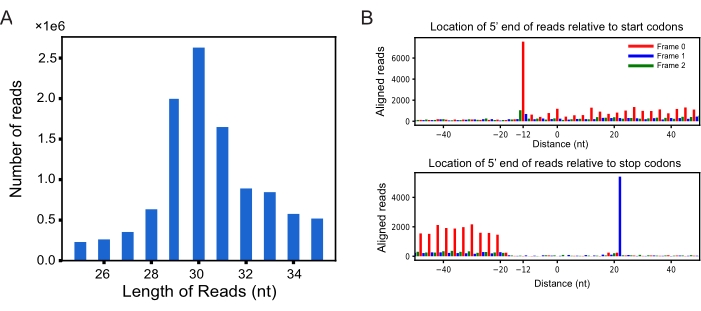
Figure 1: Assessment of sequencing reads and the P-site positions. (A) Length distribution of ribosome protected fragments (RPFs) in EIF3E-deficient cells in replicate 1 (si-eIF3e-1); (B) Inferring P-site position of RPFs of 29nt based on their densities around the known start (top) and stop codons (bottom). Please click here to view a larger version of this figure.
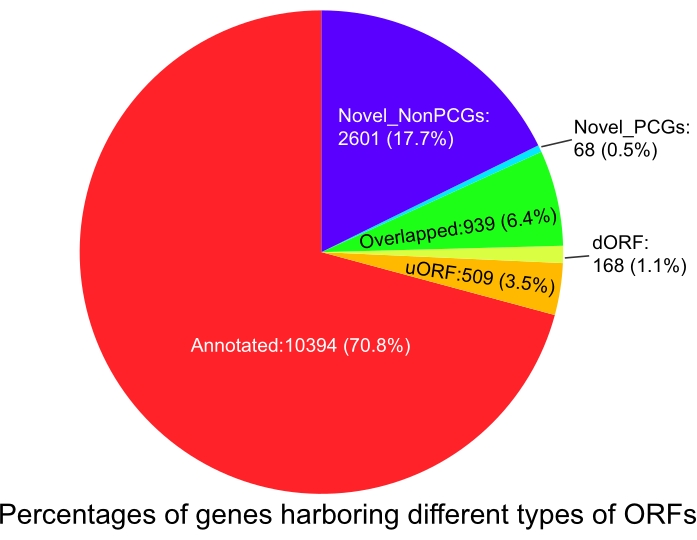
Figure 2: Percentages of genes harboring different types of ORFs identified by RiboCode using all samples together. Abbreviations: ORF = open reading frame; dORF = downstream ORF; PCG = protein-coding gene; NonPCG = nonprotein-coding gene; uORF = upstream ORF. Please click here to view a larger version of this figure.
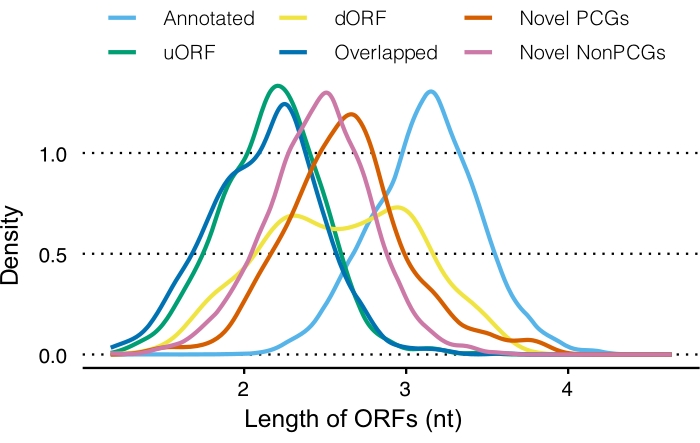
Figure 3: Length distributions of different ORF types. Abbreviations: ORF = open reading frame; dORF = downstream ORF; PCG = protein-coding gene; NonPCG = nonprotein-coding gene; uORF = upstream ORF; nt = nucleotide. Please click here to view a larger version of this figure.
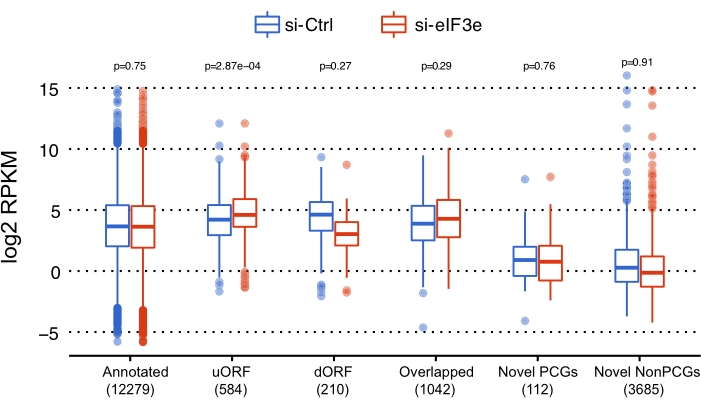
Figure 4: Comparison of normalized read counts for different ORF types between control and EIF3E-deficient cells. p-values were determined by Wilcoxon signed rank test. Abbreviation: ORF = open reading frame; dORF = downstream ORF; PCG = protein-coding gene; NonPCG = nonprotein-coding gene; uORF = upstream ORF; RPKM = Reads per kilobase per million mapped reads; siRNA = small-interfering RNA; si-Ctrl = control siRNA; si-eIF3e = siRNA targeting EIF3E. Please click here to view a larger version of this figure.
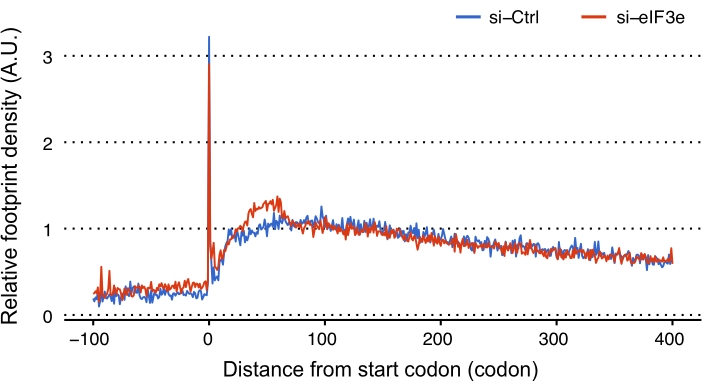
Figure 5: Metagene analysis showing the stall of ribosomes at the 25-75th codon downstream of the start codon of annotated ORFs. Abbreviation: ORF = open reading frame; siRNA = small-interfering RNA; si-Ctrl = control siRNA; si-eIF3e = siRNA targeting EIF3E; A. U., any unit. Please click here to view a larger version of this figure.
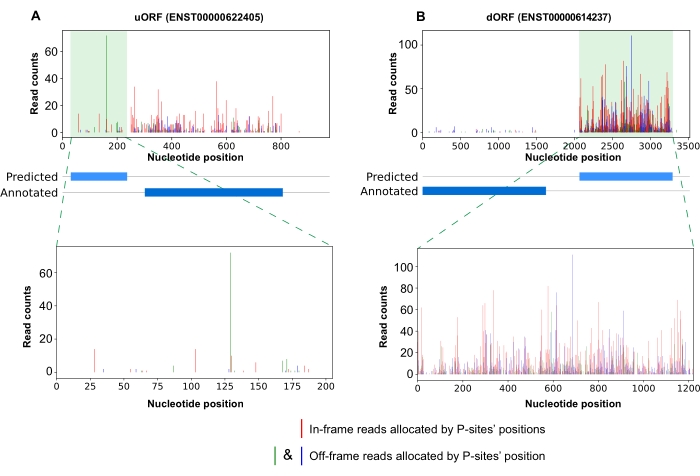
Figure 6: P-site density profiles of example ORFs encoding micropeptides. (A) P-site densities of predicted uORF and its position relative to annotated CDS on transcript ENST00000622405; (B) same as in A but for the predicted dORF on transcript ENST00000614237. Bottom panel showing the enlarged view of predicted uORF (A) or dORF (B). Red bar = in-frame reads; Green & blue bars = off-frame reads. Abbreviation: ORF = open reading frame; dORF = downstream ORF; uORF = upstream ORF; CDS = coding sequences. Please click here to view a larger version of this figure.
Supplemental Information: Evaluation of the dependence between two p-values and explanation of RiboCode results (uORF of ATF4 as an example). Please click here to download this File.
Supplemental File 1: The configuration file for RiboCode defining the selected lengths of RPFs and P-site positions. Please click here to download this File.
Supplemental File 2: RiboCode output file containing the information of predicted ORFs. Please click here to download this File.
Supplemental File 3: R script file for performing basic statistics of RiboCode output. Please click here to download this File.
Supplemental File 4: The configuration file (for RiboMiner) modified from Supplemental File 1. Please click here to download this File.
