Independent replicates reveal reproducible hits
To illustrate the expected variation between the replicate screens, the means and variation in sample mobility are plotted from three representative plates from a recent screen (Figure 1). Three replicates of the screen were performed on three different days. All three plates have negative (solvent-only) controls (darker bars), and samples within the set contained established nematicides. The remaining compounds are from a custom library currently being characterized in the Roy lab. Compared to similar screens done with C. elegans with the same molecules (SC, JK, PJR, unpublished results), the hit rate with D. dipsaci is significantly lower, and many drug plates exhibit no activity (Figure 1A). Some plates have fully reproducible hits (Figure 1B,C), while others vary in activity (Figure 1C). Relative to other species screened (not shown), D. dipsaci shows less variability in its response to compounds. Regardless, replicates are considered necessary in the identification of reproducible hits.
Nematicides vary in their ability to immobilize Ditylenchus dipsaci
Of the seven characterized nematicides tested against D. dipsaci, only Fluopyram exhibits robust activity in the assay described herein (Figure 1C). This is consistent with previous work showing that D. dipsaci is tolerant of nematicides13,14. Fluopyram inhibits complex II of the electron transport chain in a nematode-selective manner and is a commercial nematicide used to control a variety of PPNs, including Rotylenchulus reniformis and Meloidogyne incognita4,21,22. Fluopyram and one of the nematicides that lacked activity at the concentration tested (Oxamyl)23 were investigated in more detail through a dose-response analysis with D. dipsaci. Fluopyram induces an apparent dose-dependent effect on D. dipsaci mobility with an EC50 of 9.3 μM (with a 95% confidence interval between 8.2 to 10.5 μM) (Figure 2A,B). This result is expected based on published in vitro results from Storelli et al., 202013. Oxamyl has no significant effect on mobility up to a concentration of 120 µM (p = 0.3632, unpaired t-test) (Figure 2C,D).
Sodium hydroxide improves assay sensitivity
In contrast to the near-continuous swimming activity of C. elegans in liquid culture18, D. dipsaci animals are dramatically less mobile. This is not uncommon among parasitic nematodes in culture16. To help distinguish 'resting' worms from sick worms, 40 mM of NaOH is used at the assay endpoint to stimulate movement in those individuals who are capable (Figure 3)19,20. This technique allows for the clear identification of small molecules that immobilize worms, given that all worms in negative control wells move and yield an exceptionally low background of false positives in the screen.
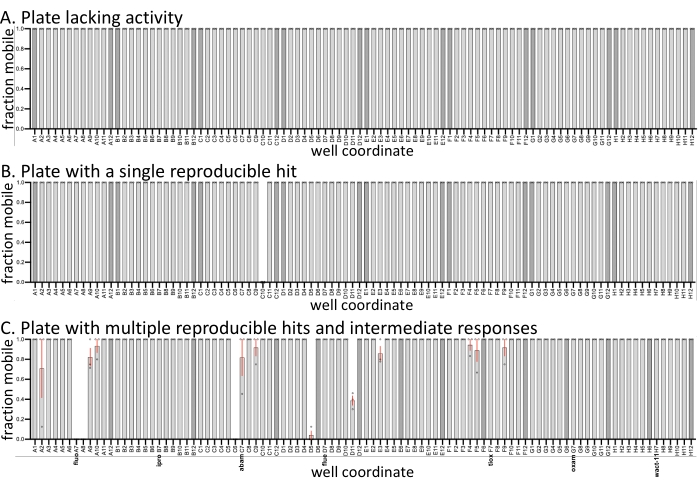
Figure 1: Examples of screen output. Three biological replicates (open circles) with D. dipsaci were performed against the small molecules (60 µM) in each of the three plates shown. All three plates have 0.6% of DMSO solvent-only controls (darker bars). Except where otherwise noted with solvent controls or known nematicides, the wells of the three plates contain relatively uncharacterized drug-like compounds purchased from vendors (see Burns et al., 2015)18. (A) A 96 well plate from the small molecule library lacks any molecule with observable bioactivity against D. dipsaci. (B) A 96 well plate from the small molecule library has a single molecule that reproducibly disrupts D. dipsaci mobility. (C) A 96 well drug plate containing the characterized nematicides fluopyram (fluo), iprodione (ipro), abamectin (abam), fluensulfone (flue), tioxazafen (tiox), oxamyl (oxam), and wact-11. The error bars represent the standard error of the mean. Please click here to view a larger version of this figure.
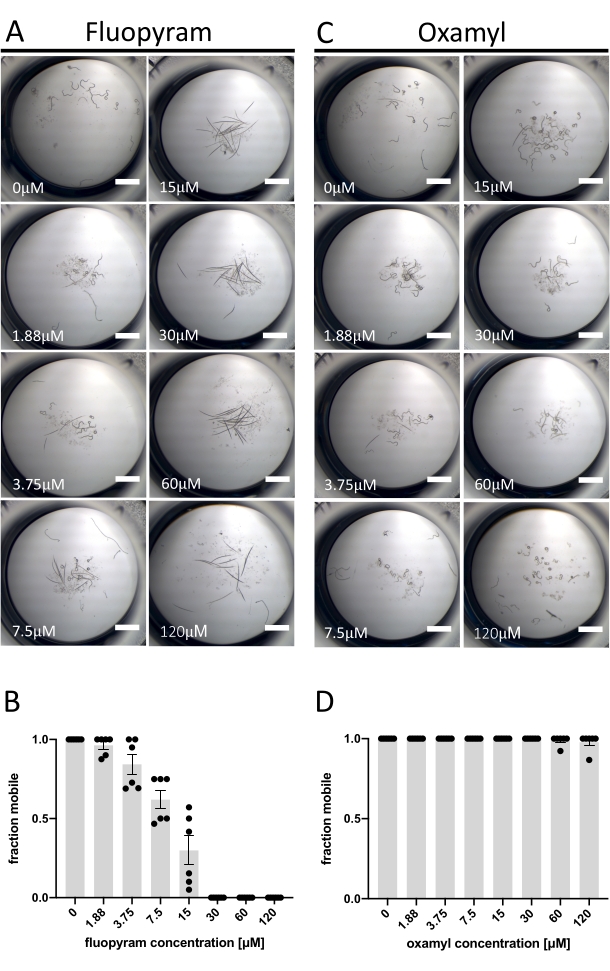
Figure 2: Example of positive and negative results using mobility assay. (A) Examples of the terminal phenotypes after the exposure of D. dipsaci to increasing concentrations of fluopyram after 5 days before addition of NaOH. (B) A dose-response analysis of the movement of D. dipsaci after 5 days of exposure to the indicated concentrations of fluopyram. (C,D) The same as A,B, except for oxamyl. Each graph shows trials done on two separate days with three replicates each day. The y-axis of each graph indicates the fraction mobile of the worms in each well calculated as the number of animals moving relative to the total number of animals in the well. The error bars on both graphs represent the standard error of the mean. The scale bar represents 1 mm. Please click here to view a larger version of this figure.
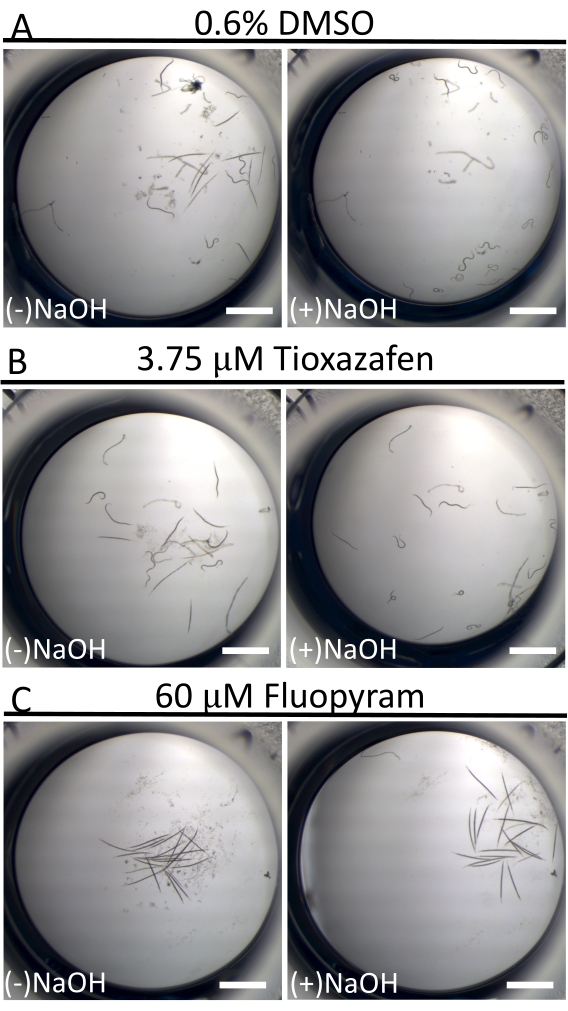
Figure 3: Mobility of D. dipsaci after adding 40 mM of NaOH. The effect of NaOH addition to D. dipsaci samples in the presence of solvent-only control (A), tioxazafen (B), or fluopyram (C) after 5 days of co-incubation. The scale bar represents 1 mm. Please click here to view a larger version of this figure.
