Protein phosphorylation controls most cellular processes, including but not limited to the response to DNA damage, growth factor signaling, and the passage through mitosis1,2,3. In mammalian cells, the majority of proteins are phosphorylated at one or more serine, threonine, or tyrosine residues at some point in time, with phosphoserines and phosphothreonines comprising approximately 98% of all phosphorylation sites2,3. While kinases have been extensively studied in cellular signaling, the role of PPPs in the regulation of dynamic cellular processes is still emerging.
Phosphorylation dynamics are controlled by the dynamic interplay between kinases and phosphatases. In mammalian cells, there are more than 400 protein kinases that catalyze serine/threonine phosphorylation. Over 90% of these sites are dephosphorylated by phosphoprotein phosphatases (PPPs), a small family of enzymes that consists of PP1, PP2A, PP2B, PP4-7, PPT, and PPZ2,3. PP1 and PP2A are responsible for the majority of phosphoserine and phosphothreonine dephosphorylation within a cell2,3,4. The notable difference in number between kinases and phosphatases and the lack of specificity of PPP catalytic subunits in vitro led to the belief that kinases are the main determinant of phosphorylation2,3. However, multiple studies have shown phosphatases to establish substrate specificity through the formation of multimeric holoenzymes5,6,7,8,9. For example, PP1 is a heterodimer that consists of a catalytic subunit and, at a given time, one out of the more than 150 regulatory subunits6,7,8. Conversely, PP2A is a heterotrimer that is formed of a scaffolding (A), a regulatory (B), and a catalytic (C) subunit2,3,9. There are four distinct families of PP2A regulatory subunits (B55, B56, PR72, and striatin), each with multiple genes, splice variants, and localization patterns2,3,9. The multimeric nature of PPPs fills the gap in the number of kinases and PPP catalytic subunits. However, it creates analytical challenges for studying PPP signaling. To comprehensively analyze PPP signaling, it is critical to investigate the various holoenzymes within a cell or tissue. Great advances have been made in studying the human kinome through the use of kinase inhibitor beads, termed multiplex inhibitor beads or kinobeads, a chemical proteomic strategy where kinase inhibitors are immobilized on beads and mass spectrometry is used to identify enriched kinases and their interactors10,11,12,13.
We have established a similar approach to study PPP biology. This technique involves affinity capture of PPP catalytic subunits using beads with an immobilized, non-selective PPP inhibitor called microcystin-LR (MCLR) termed phosphatase inhibitor beads (PIBs)14,15. Unlike other methods that require the endogenous tagging or expression of exogenous PPP subunits that could alter protein activity or localization, PIB-MS allows for the enrichment of endogenous PPP catalytic subunits, their associated regulatory and scaffolding subunits, and interacting proteins (termed the PPPome) from cells and tissues at a given time point or under specific treatment conditions. MCLR inhibits PP1, PP2A, PP4-6, PPT, and PPZ at nanomolar concentrations, making PIBs highly effective at enriching for the PPPome16. This method can be scaled for use on any starting material from cells to clinical samples. Here, we describe in detail the use of PIBs and mass spectrometry (PIB-MS) to efficiently capture, identify, and quantify the endogenous PPPome and its modification states.
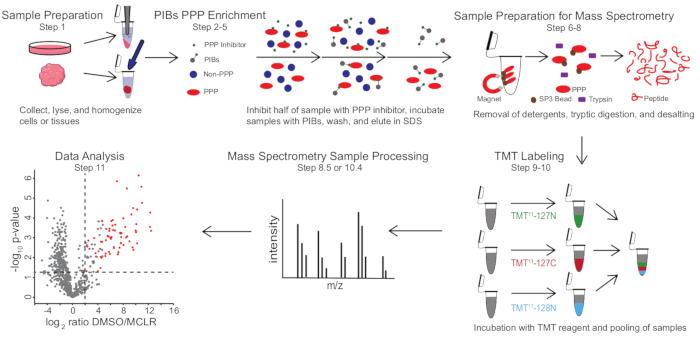
Figure 1: Visual summary of the PIB-MS protocol. In a PIB-MS experiment, samples can be obtained in various forms, from cells to tumors. The sample is collected, lysed, and homogenized prior to PPP enrichment. To enrich for PPPs, the lysate is incubated with PIBs with or without a PPP-inhibitor, such as MCLR. The PIBs are then washed, and PPPs are eluted in denaturing conditions. The samples are prepared for mass spectrometry analysis by the removal of detergents through SP3 protein enrichment, tryptic digestion, and desalting. Samples can then be optionally TMT-labeled prior to mass spectrometry analysis. Please click here to view a larger version of this figure.
PIB-MS involves lysis and clarification of cells or tissues, incubation of the lysate with PIBs, elution, and analysis of the eluate via western blotting or mass spectrometry-based approaches (Figure 1). The addition of free MCLR can be used as a control to distinguish specific PIB binders from non-specific interactors. For most applications, a label-free approach can be used to directly identify proteins in eluates. In cases where greater precision in quantification or the identification of low-abundance species is needed, further processing with tandem mass-tag (TMT) labeling can be used to increase coverage and decrease input.
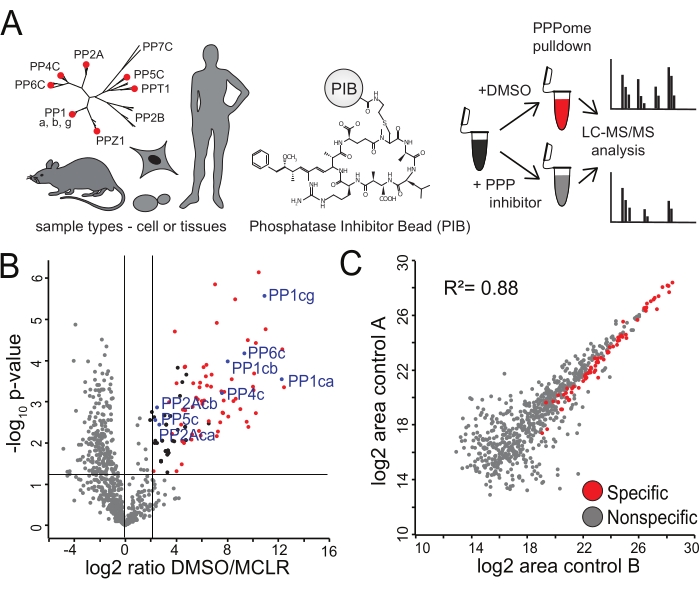
Figure 2: Identification of specific PIBs binders. (A) A variety of tissue types or cells can be analyzed via PIB-MS. HeLa cells in biological triplicate were either treated with DMSO or the PPP-inhibitor MCLR, incubated with PIBs, and analyzed via LC-MS/MS. (B) Volcano plot of PIB-MS analysis in DMSO and MCLR-treated HeLa cell lysates, with specific PIB binders shown in red. PPP catalytic subunits are labeled and shown in blue. New candidate PPP subunits or interacting proteins are shown in black. Non-specific binders are shown in grey. (C) Scatter plot of log2 area of two biological replicates from HeLa cell lysates treated with DMSO to demonstrate the reproducibility of the enrichment. Specific binders to PIBs are shown in red. Please click here to view a larger version of this figure.
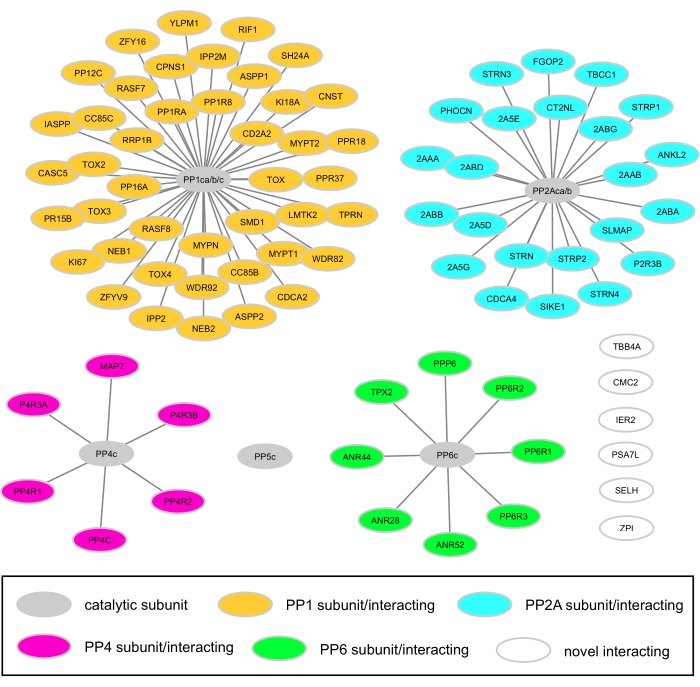
Figure 3: Network analysis of all PPP subunits and PPP-interacting proteins identified in HeLa cells. These proteins were found to be specific PIB interactors in the HeLa PIB pulldown with and without MCLR. Please click here to view a larger version of this figure.
To demonstrate the performance of the PIBs, we performed a PIB pulldown experiment in HeLa cells to identify PPPs and their interactors (Figure 2A). HeLa cells were grown in biological triplicate and lysed. Each triplicate lysate was split in half, where half was treated with MCLR for 15 min to prevent the binding of PPP catalytic subunits or DMSO before PIB enrichment was performed. Following PIB enrichment, a label-free comparison of the abundance of proteins quantified in the DMSO-treated and MCLR-treated samples was performed to distinguish specific from non-specific interactors (Figure 2B). In the analysis, we detected all MCLR-sensitive PPP catalytic subunits PP1ca, PP1cb, PP1cc, PP2Aca, PP2Acb, PP4c, PP5c, and PP6c. In the DMSO-treated samples, the abundances (log2 areas of protein quantification) were highly correlated, indicating reproducibility (Figure 2C), yet upon treatment with MCLR, the PPP catalytic subunit or PPP interacting protein abundance in PIB pulldowns was greatly reduced (Figure 2B). In this analysis, we identified 92 PPP subunits and specific PPP interactors (see Supplementary Table 1), which is comparable to a previous PIB-MS analysis in HeLa cells14 (Figure 3).
Supplementary Table 1: MS data of representative PIB-MS analysis. Please click here to download this Table.
