Here we will use the validated microprotein mitoregulin (Mtln) as an example to demonstrate how a conserved sORF will generate a positive PhyloCSF score that can be easily visualized and analyzed on the UCSC Genome Browser. Mitoregulin was previously annotated as a noncoding RNA (formerly human gene ID LINC00116 and mouse gene ID 1500011K16Rik). Comparative genomics and sequence conservation analysis methods played a critical role in its initial discovery40,57,58,59,60,61, highlighting the strength of these methods. For this example, the mouse GRCm38/mm10 (Dec. 2011) assembly will be used. The search can be performed using the gene identifiers (mitoregulin, Mtln) or the gene position (chr2:127,791,364-127,792,496) as described in protocol section 2. Alternatively, the amino acid sequence for mitoregulin (shown in Figure 2) can be searched using the BLAT tool (described in protocol section 3).
A screen similar to the one depicted in Figure 1A will appear with the PhyloCSF Track Hub visible at the top of the screen. The Smoothed PhyloCSF tracks (smoothed with a hidden Markov model defining a probability that each codon is coding) are depicted as six total tracks, with three tracks corresponding to the plus strand of DNA (depicted in green as PhyloCSF +1, +2 and +3) and three tracks corresponding to the minus strand of DNA (depicted in red as PhyloCSF -1, -2 and -3). These tracks represent the three potential reading frames for the gene of interest in each direction. On the browser window, exons are depicted as blue rectangles connected by thin blue horizontal lines, which represent the introns. The arrowheads on the intronic regions indicate which direction the gene is transcribed in (and thus, which strand to focus on for the PhyloCSF score). For the example of Mtln de Figure 1, the intronic arrowheads are pointing to the left. Therefore, the Mtln gene is transcribed from the minus strand of DNA, and the relevant PhyloCSF score is depicted in the -1, -2, and -3 tracks (in red).
Each PhyloCSF track is depicted as a thin black line with negative scoring regions depicted in light green/red below the line and positive scoring regions indicated in dark green/red above the line. As described in the introduction, a positive PhyloCSF score indicates a conserved region that is likely coding. Note that for protein-coding regions with particularly high sequence conservation, they often also score positively on the antisense strand; however, the PhyloCSF score is usually higher on the correct strand. For example, this can be seen in Figure 1 for Mtln where the correct coding sequence scores very highly in the PhyloCSF -1 track, and the antisense strand (PhyloCSF +2 track) also generates a positive score. As seen in Figure 1A (indicated with black box), there is a region in the first exon of Mtln that scores very highly on the PhyloCSF -1 track, suggesting this may correspond to a coding region. To examine this region in further detail, it is helpful to zoom in and magnify the region (Figure 1B). As shown in Figure 1C,D, the positively scoring region in the first exon of Mtln begins directly over a start codon (Figure 1C) and terminates at a stop codon (Figure 1D), which indicates this ORF is highly conserved and strongly suggests it is a coding ORF. As Mtln is on the minus strand of DNA, the start and stop codons are shown as the reverse complement of the codon (i.e., the ATG start codon is shown as CAT [Figure 1C] and the TGA stop codon is shown as TCA [Figure 1D]).
In addition to using PhyloCSF to search for conserved regions with microprotein-coding potential, this technique can also be applied as a first-pass analysis of putative noncoding RNAs to rule out the presence of a conserved ORF, thus providing support for a noncoding annotation. For example, analysis of the well-characterized lncRNA HOTAIR62,63 using PhyloCSF shows a negative score throughout the entire gene across all six tracks (Figure 3), strongly indicating a lack of sequence conservation and providing support that HOTAIR is correctly annotated as a noncoding RNA.
As clearly seen in Figure 1, the entire coding ORF for mitoregulin is located within a single exon, thereby producing a simple and straightforward readout by PhyloCSF with a single, uninterrupted, positively scoring region. However, PhyloCSF track hub data is not always as clear-cut and easy to interpret. For example, the mitolamban/Stmp1/Mm47 microprotein encoded by the mouse 1810058I24Rik gene47,64,65 depicts a conserved ORF that spans three exons (Figure 4A), and the positive PhyloCSF score jumps from the +2 track in exon 1 (Figure 4B) to the +3 track in exon 2 (Figure 4C), and then back to the +2 track in exon 3 (Figure 4D). While at first glance this looks confusing, the explanation is quite straightforward. PhyloCSF scores the six potential reading frames (three on the plus strand of DNA and three on the minus strand) of genomic regions without considering the specific exon/intron architecture for each gene. Therefore, it retains the intronic sequence information in the 3-nucleotide periodicity of the reading frames. Thus, if an intron contains a number of nucleotides that is not divisible by three (i.e., three nucleotides/codon), the PhyloCSF reading frame will jump from one track to another.
Lastly, PhyloCSF can also be effectively used to identify multiple distinct coding ORFs within a single RNA molecule. For example, the MIEF1 microprotein (MIEF1-MP) is encoded within the 5' UTR of mitochondrial elongation factor 1 (MIEF1)66 (Figure 5). When the MIEF1 genomic region is analyzed by PhyloCSF, a discrete positive PhyloCSF score corresponding to the MIEF1-MP (Figure 5C) can be readily observed upstream of the main CDS for MIEF1 (Figure 5B). Further discussion on MIEF1 and its associated microprotein (MIEF1-MP) is provided below in the discussion along with a summary of the strengths and weaknesses of the methods and protocols outlined in this article.

Figure 1: PhyloCSF analysis of the mitoregulin (Mtln) gene indicates a region of high sequence conservation corresponding to a validated microprotein. (A) Screenshots of the UCSC Genome Browser and PhyloCSF Tracks show that Mtln contains two exons and a single intron. The arrowheads within the intron point to the left, indicating the Mtln gene is transcribed from the minus strand of DNA, and the relevant PhyloCSF scores are therefore shown in the -1, -2, and -3 tracks (in red). The complete mitoregulin coding sequence is contained within Exon 1 and scores highly on the PhyloCSF -1 track (B). A conserved start codon can be clearly observed at the beginning of the positively scoring region in the PhyloCSF -1 track (C), which is highlighted with a green box (CAT, reverse complement ATG). Additionally, a conserved stop codon (TCA, reverse complement TGA) is indicated with a red box in panel (D), which aligns with the end of the positively scoring PhyloCSF region. Detailed information about the Mtln gene can be found by clicking on the Mtln gene identifier within the blue box (shown in panel A). Of note, highly conserved protein-coding regions often also score positively on the antisense strand (seen here in the PhyloCSF +2 track for Mtln). However, the PhyloCSF score is typically higher on the correct strand (the PhyloCSF -1 track in this example). Please click here to view a larger version of this figure.

Figure 2: Multiple species sequence alignment of the microprotein mitoregulin generated using the Clustal Omega program. The mitoregulin amino acid sequences for the eight species indicated were extracted as detailed in protocol section 6 and aligned with the Clustal Omega multiple sequence alignment tool. The properties of the amino acids are indicated by color (red, small/hydrophobic; blue, acidic; magenta, basic; green, hydroxl/sulfhydryl/amine) (further defined in Table 2). The symbols below the amino acids indicate the degree of conservation (asterisks, fully-conserved residues; colons, amino acids with strongly similar properties; periods, conservation between groups of weakly similar properties) (detailed extensively in Table 1). Please click here to view a larger version of this figure.
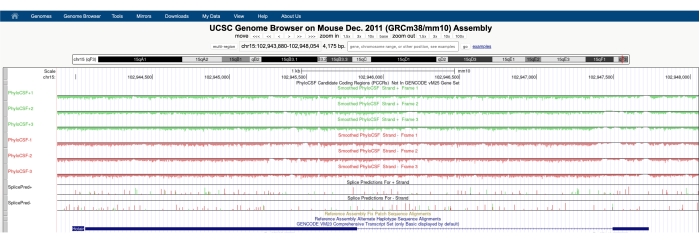
Figure 3: A screenshot of the PhyloCSF tracks for the validated long noncoding RNA Hotair shows a lack of sequence conservation throughout its genomic locus. The arrowheads in the intronic region of Hotair are pointing left, indicating that the lncRNA is transcribed from the negative strand of DNA, and therefore the PhyloCSF -1, -2, and -3 tracks should be the focus of analysis. Note that the PhyloCSF score is negative throughout the entire gene (for all six tracks), indicating a lack of sequence conservation, which supports its proper annotation as a noncoding RNA. Please click here to view a larger version of this figure.
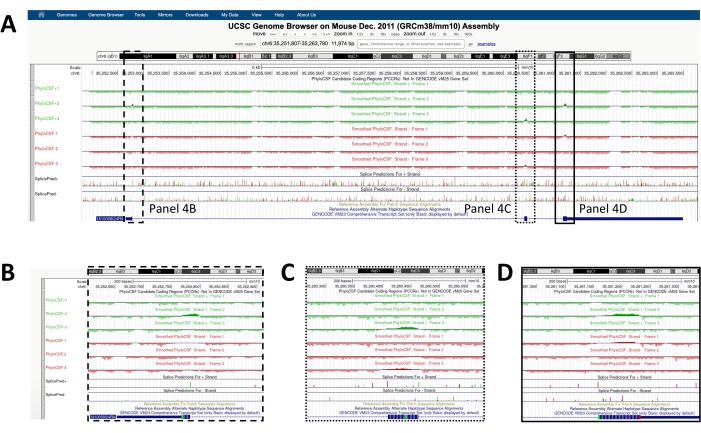
Figure 4: PhyloCSF analysis of the mouse 1810058I24Rik gene, which encodes the microprotein mitolamban/Stmp1/Mm47. (A) The mouse 1810058I24Rik gene is comprised of three exons, and the arrowheads in the intronic regions point right, indicating it is transcribed on the plus strand of DNA and therefore the PhyloCSF +1, +2, and +3 tracks should be analyzed. The conserved microprotein coding sequence spans all three exons, starting in exon 1 (B), reading through exon 2 (C), and ending in exon 3 (D). Note that the positive PhyloCSF score is found on the +2 track in exon 1, the +3 track in exon 2, and the +2 track in exon 1. The reason for the movement of the positive score from one track to the other is that PhyloCSF analyzes the six potential reading frames of the DNA sequence independent of the gene's exon/intron structure. Therefore, an intron containing a number of nucleotides that is not divisible by three (three nucleotides/codon) will cause a shift in the reading frame to a different track. Please click here to view a larger version of this figure.

Figure 5: Analysis of the Mief1 genomic locus with PhyloCSF identifies a region with protein-coding potential in the 5' UTR that is independent of the main Mief1 CDS on the shared RNA. This conserved upstream ORF (uORF) has been shown to encode a microprotein named Mief1-MP. (A) Overview of the Mief1 genomic locus. The arrowheads in the introns point to the right, indicating Mief1 is transcribed from the plus strand of DNA (focus on the PhyloCSF +1, +2, and +3 tracks to determine coding potential). The main Mief1 CDS encodes a 463 amino acid protein and is shown in panel (B). However, there is also a distinct conserved upstream ORF within the 5' UTR of Mief1 that encodes a unique 70 amino acid microprotein called Mief1-MP (C). As seen in Panel C, the Mief1-MP has its own conserved start and stop codon within the Mief1 5' UTR, and the ORF scores very highly on the PhyloCSF +1 track, providing strong evidence that it encodes a functional microprotein. Abbreviations: ORF = open reading frame; uORF = upstream ORF; UTR = untranslated region; CDS = coding sequence. Please click here to view a larger version of this figure.
| Symbol | Level of Amino Acid Conservation | Grouped Amino Acids |
| Asterisk (*) | Fully-conserved residue | Not applicable (single, fully-conserved residue) |
| Colon (:) | Groups with strongly similar properties | STA; NEQK; NHQK; NDEQ; QHRK; MILV; MILF; HY; FYW |
| Period (.) | Groups with weakly similar properties | CSA; ATV; SAG; STNK; STPA; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; HFY |
| Space (no symbol) | No similarity | Not applicable (no similarity) |
Table 1: Definitions of consensus symbols for Multiple Sequence Alignments generated by Clustal Omega. The multiple species sequence alignment shown in Figure 2 was generated using Clustal Omega52. Abbreviations: serine (S), threonine (T), alanine (A), asparagine (N), glutamic acid (E), glutamine (Q), lysine (K), aspartic acid (D), arginine (R), methionine (M), isoleucine (I), leucine (L), phenylalanine (F), histidine (H), tyrosine (Y), tryptophan (W), cysteine (C), valine (V), glycine (G), proline (P).
| Font Color | Property | Amino Acid Residue [Abbreviation] |
| Red | Small, hydrophobic | alanine [A], valine [V], phenylalanine [F], proline [P], methionine [M], isoleucine [I], leucine [L], tryptophan [W] |
| Blue | Acidic | aspartic acid [D], glutamic acid [E] |
| Magenta | Basic | arginine [R], lysine [K] |
| Green | Hydroxl, sulfhydryl, amine, +G | serine [S], threonine [T], tyrosine [Y], histidine [H], cysteine [C], asparagine [N], glycine [G], glutamine [Q] |
Table 2: Properties of the amino acids depicted in Figure 2. Clustal Omega52 was used to generate the multiple sequence alignment shown in Figure 2.
