Integrating PLGA fibers, dimple embedding, and agitation leads to a robust generation of cerebral organoids that enables iPSC-derived cultures to be maintained for extended periods of time (Figure 1).
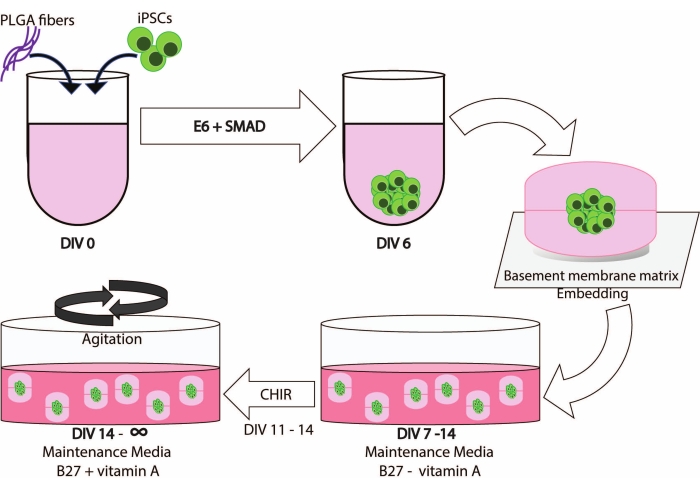
Figure 1: Schematic illustration of the workflow and timeline of this method. Please click here to view a larger version of this figure.
The initial neural induction period is comparable to previously published procedures11. The embryoid bodies (EB) begin as circular aggregates (Figure 2B) with white or transparent edges. As the EB is changed from neural induction media to neural maintenance media at DIV7, protrusions and buddings emerge within 24 h from the circular tissue (Figure 2C). Furthermore, as the organoid continues to grow, the surface area of the PLGA fiber helps the organoid to elongate (Figure 2D). During maturation, the edges of the organoid need to remain intact, as it is a good sign of healthy cells and development; otherwise, additional nutrients must be provided13 (Figure 2E). Growth of the organoids is further facilitated by the agitation of the organoid culture, as this improves perfusion with nutrients.
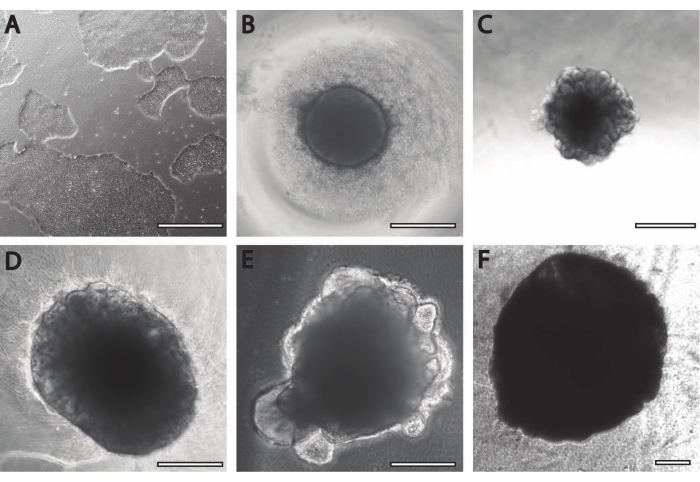
Figure 2: Differentiation and maturation of cerebral organoids. (A) Representative image of an iPSC culture at 70%-80% confluency. (B) Embryoid bodies (EBs) were generated and neuroepithelial formation was induced until DIV7. EBs were then embedded in the membrane matrix and further differentiated toward cerebral organoids. (C) Organoids at DIV10 displaying distinct budding formations. Organoids can be further matured in the membrane matrix using either (D) dimple or (E) sandwich embedding, here shown at DIV30. (F) Long-term culture shows cerebral organoids growing to significant sizes (DIV70). Scale bar = 500 µm. Please click here to view a larger version of this figure.
The dimensions and external morphology of the organoid are complemented by a complex architecture inside of the organoid. After fixing the tissue at DIV7 after the first stage of differentiation, cells of the EB express SOX2, an HMG box transcription factor that acts as a marker for multipotent neural stem cells21, as well as paired protein box Pax-6, indicating neural progenitor cells (Figure 3). Immature neurons marked by TuJ1 (class III β-tubulin)22 can already be seen scattered throughout the tissue.
At this stage, an example of the self-organization that these organoids go through becomes apparent. The organization of radial structures, called rosettes, is analogous to the neural tube, with SOX2+ cells in the rosette center21 and PAX6 towards the rosette periphery. These rosettes give rise to neurons as they migrate out. These radiating cells are initially double positive for the neural stem/progenitor cell marker Nestin23 and glial fibrillary acidic protein (GFAP), similar to the radial glia found in neurogenic areas of the in vivo brain. As these migrating neurons mature, the cytoskeletal markers reflect this change22. The early-stage neural differentiation marker TuJ122 is visible in the inner circle of the rosette and shifts to microtubule-associated protein 2 (MAP2)24, a neuron-specific maturation marker at the periphery.
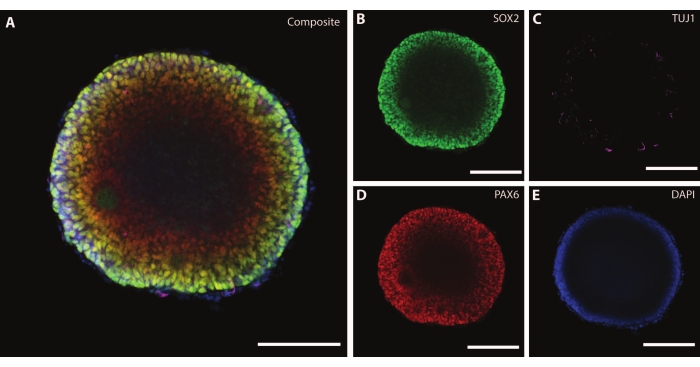
Figure 3: Embryoid bodies at DIV7 show structural organization and immature characterization. Whole mount immunohistochemistry image of an EB at DIV7. (A) A composite image of the EB displaying (B) SOX2+ neural rosettes, (C) immature neurons (TUJ1) scattered throughout the EB, (D) neural progenitor cells (PAX6), and (E) nuclei visualized by DAPI. Scale bar = 100 µm. Please click here to view a larger version of this figure.
As the organoids age, the organization and markers of the developing neurons start replicating physiological conditions. At DIV30, many rosettes giving rise to neurogenic regions equivalent to the developing brain can be observed25. By DIV60, these SOX2+ neurogenic regions are non-existent and are replaced by mature MAP2 and NeuN26, a neuron differentiation marker, and positive neurons (Figure 4 and Figure 5).
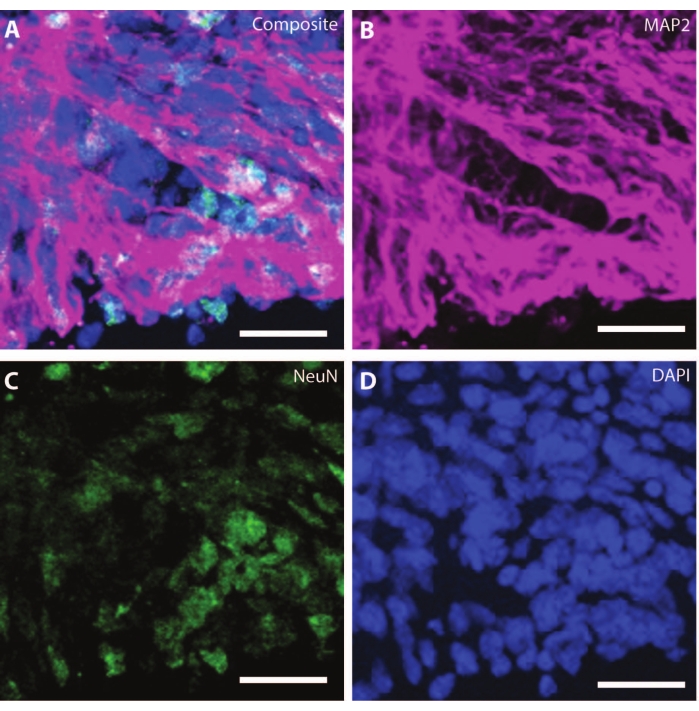
Figure 4: Organoids at DIV120 show mature neuronal characterization. Immunohistochemistry image of an organoid section at DIV 120. (A) A composite image of the section showing mature neuronal markers of (B) MAP2 (purple) and (C) NeuN (green). (D) Nuclei were visualized by DAPI. Scale bar = 20 µm. Please click here to view a larger version of this figure.
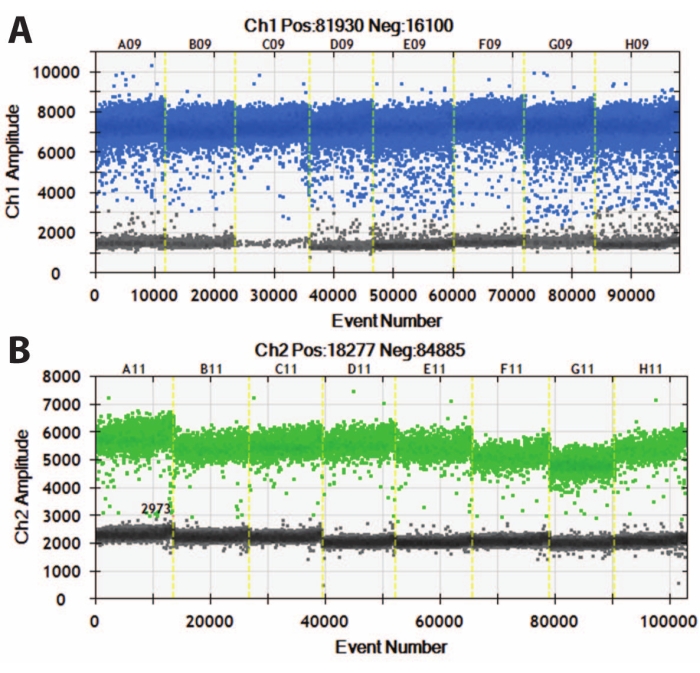
Figure 5: Digital droplet PCR of DIV120 organoids. Digital droplet PCR graphs showing the absolute expression value of (A) MAP2 (top, blue) and (B) NeuN (bottom, green). Slashed yellow lines separate different iPSC lines (A, B, C, etc.), and organoids have been batched together. N = 5. Please click here to view a larger version of this figure.
These cytoskeletal markers can be used in conjunction with other post-mitotic markers (like doublecortin27 and synapsin28) to probe for synaptic plasticity and other age-related declines (Figure 6), as well as additional brain tissue like astrocytes and glia (Figure 7).
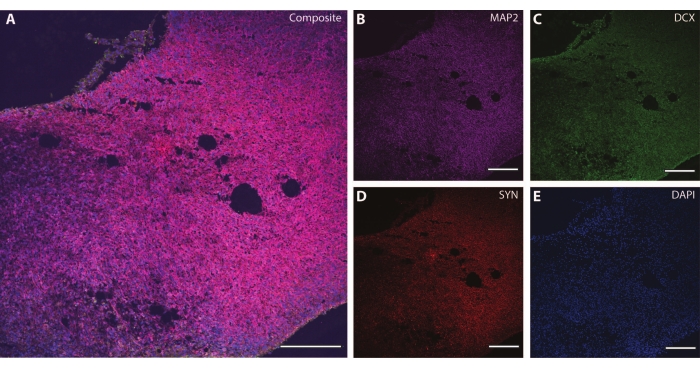
Figure 6: Example of synaptic terminal analysis. Immunohistochemistry image of a section of the organoid at DIV 120. (A) A composite image of the section stained for (B) MAP2, (C) doublecortin (DCX), and (D) synapsin I (SYN). (E) Nuclei were visualized by DAPI. Scale bar = 200 µm. Please click here to view a larger version of this figure.
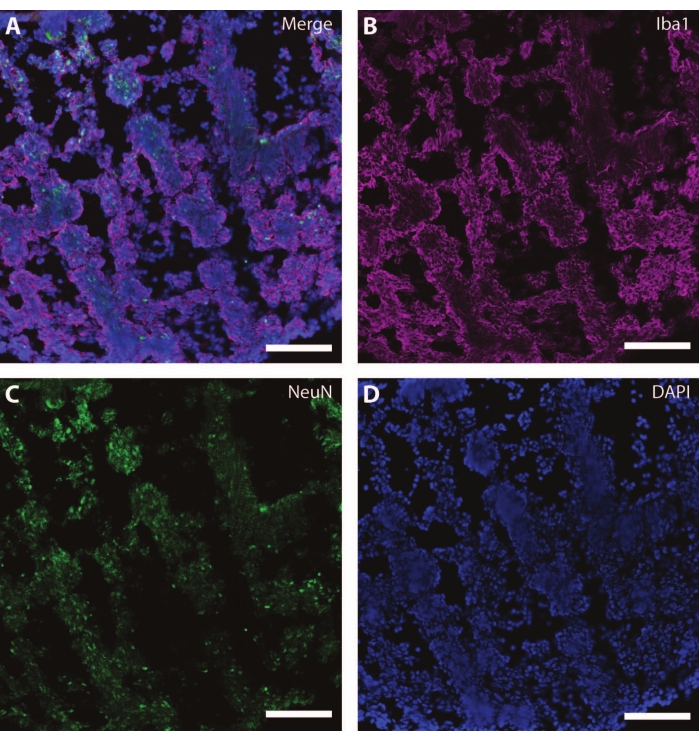
Figure 7: Evidence of glial development. Immunohistochemistry image of a section of the organoid at DIV 120. (A) A composite image of the section stained for (B) ionized calcium-binding adaptor molecule 1 (Iba1) and (C) NeuN. (D) Nuclei were visualized by DAPI. Scale bar = 100 µm. Please click here to view a larger version of this figure.
| Reagent | Final Concentration | Volume (50 mL total) |
| DMEM-F12 | 50% | 25 mL |
| Neurobasal Medium | 50% | 25 mL |
| N2 Supplement (100x) | 1x | 0.25 mL |
| B27 Supplement -/+ Vitamin A (50x) | 0.5x | 0.5 mL |
| Insulin | 0.25% | 12.5 µL |
| GlutaMAX (100x) | 1x | 0.5 mL |
| MEM-NEAA (100x) | 0.5x | 0.25 mL |
| HEPES (1 M) | 10 mM | 0.5 mL |
| Antibiotic/Antimycotic (100x) | 1x | 0.5 mL |
| 2-β-mercaptoethanol | 50 µM | 17.5 µL |
| *NOTE: some DMEM-F12 already contains GlutaMAX, no need to add additional. | ||
Table 1: Composition of the differentiation media used in the present study.
