Immunoprecipitation with an Anti-Epitope Tag Affinity Gel to Study Protein-Protein Interactions
Summary
Protein-protein interactions are important for elucidating the function of target proteins, and co-immunoprecipitation (co-IP) can easily confirm PPIs. We transiently transfected a plasmid encoding an epitope-tagged protein into HEK-293 cells and developed an immunoprecipitation method to easily confirm the binding of two target proteins.
Abstract
Protein-protein interactions (PPIs) play a pivotal role in biological phenomena, such as cellular organization, intracellular signal transduction, and transcriptional regulation. Therefore, understanding PPIs is an important starting point for further investigation of the function of the target protein. In this study, we propose a simple method to determine the binding of two target proteins by introducing mammalian expression vectors into HEK-293 cells using the polyethylenimine method, lysing the cells in homemade protein lysis buffer, and pulling down the target proteins on an epitope tag affinity gel. In addition, the PPI between the various epitope tag fused proteins can be confirmed by using affinity antibodies against each tag instead of the epitope tag affinity gel. This protocol could also be used to verify various PPIs, including nuclear extracts, from other cell lines. Therefore, it can be used as a basic method in a variety of PPI experiments. Proteins degrade by extended time course and repeated freeze-thaw cycles. Therefore, cell lysis, immunoprecipitation, and immunoblotting should be performed as seamlessly as possible.
Introduction
Proteins play a major role in all cellular functions, including information processing, metabolism, transport, decision-making, and structural organization. Proteins mediate their functions by interacting physically with other molecules. Protein-protein interactions (PPIs) are important for mediating cellular functions, such as mediating signal transduction, sensing the environment, converting energy into physical movement, regulating the activity of metabolic and signaling enzymes, and maintaining cellular organization1. Thus, PPIs can be used to elucidate unknown functions2. Methods for detecting PPIs can be classified into three types: in vitro, in vivo, and in silico. Co-immunoprecipitation (co-IP), affinity chromatography, tandem affinity purification, protein arrays, phage display, protein fragment complementation, X-ray crystallography, and nuclear magnetic resonance spectroscopy have been used for in vitro PPI detection3. Among these methods, co-IP is widely used because of its simplicity.
The fusion tag FLAG consists of eight amino acids (AspTyrLysAspAspAspAspLys: DYKDDDDK), including an enterokinase cleavage site, and was specifically designed for immunoaffinity chromatography4. DYKDDDDK-tagged proteins are recognized and captured using an anti-DYKDDDDK antibody. Therefore, they are efficiently pulled down using DYKDDDDK binding agarose beads5 to confirm their binding to specific proteins in a simple manner. Immunoprecipitation can be performed in a variety of cells, and a wide range of PPIs can be confirmed using antibodies against the protein of interest. Immunoprecipitation and peptide elution with anti-DYKDDDDK agarose beads have been previously reported5.
Here, we provide a simple immunoprecipitation method in which a plasmid encoding a DYKDDDDK-tagged protein is transiently introduced into HEK-293 cells to confirm the association of two proteins of interest. Certain DYKDDDDK antibodies can bind to both the N-terminus and C-terminus of the fusion proteins but not others6. Therefore, to avoid confusion, the antibody that recognizes the tag fused to both N- and C-terminus should be chosen. When inserting an epitope tag, it may be possible to avoid conformational changes in the protein by inserting 3 to 12 base pairs between the epitope tag and the target protein. However, the inserted sequence should be a base pair in multiples of 3 to avoid frameshift.
Protocol
Figure 1 presents an overview of the protocol.
1. Preparation of solutions and buffers
- Protein lysis buffer: Prepare the protein lysis buffer following a previously published report7 (Table 1).
- Protein lysis buffer + protease inhibitor (PI): Add protease inhibitor (see Table of Materials) to the above-prepared buffer (step 1.1). Store at -20 °C.
- Sample buffer: Mix 900 µL of 4x Laemmli sample buffer (see Table of Materials) and 100 µL of 2-mercaptoethanol. Store at -20 °C.
- Phosphate-buffered saline (PBS) with polyoxyethylene (20) sorbitan monolaurate (PBS-P): Mix 1.998 L of PBS and 2 mL of polyoxyethylene (20) sorbitan monolaurate (see Table of Materials). Store at room temperature.
- 1x Tris/Glycine/SDS buffer: Mix 450 mL of DDW and 50 mL of 10x Tris/Glycine/SDS. Prepare at room temperature on the day of use.
2. Plasmid transfection
- Culture HEK-293 cells in a 10 cm collagen-coated dish to subconfluence (80%-95%) using Dulbecco's modified eagle's medium containing 10% fetal bovine serum and 1% penicillin-streptomycin (10,000 units/mL penicillin G and 10,000 µg/mL streptomycin) (see Table of Materials).
- Prepare 1-10 µg of plasmid7 to be transfected per dish and make a transfection mix. Add 150 mM NaCl to a final volume of 250 µL per dish. Vortex and spin down the mixture.
- Add 60 µL of 1 mg/mL Polyethylenimine (PEI, see Table of Materials)per dish, followed by 150 mM NaCl to a final volume of 250 µL of PEI solution.
- Add the PEI solution to the transfection mix to make a mixed solution. Immediately vortex (at top speed for 3-5 s, perform the same thereafter) and spin down.
- Incubate at room temperature for 15-30 min. During this time, change the medium of HEK-293 cells.
- Sprinkle 500 µL/dish of mixed solution all over the dish of HEK-293 cells and incubate overnight (13-14 h).
- Perform medium exchange the next day.
3. Cell lysis and sample preparation
NOTE: To avoid protein degradation, subsequent steps should be performed without preservation or freeze-thaw of the sample as much as possible.
- Approximately 48 h after transfection, wash the cells thrice with ice-cold PBS, add 500 µL/dish of protein lysis buffer + PI, and collect the cells by cell scraper and a pipette in a tube with low protein adsorption on ice.
- Vortex every 5 min and incubate the samples on ice for 10 min.
- Centrifuge at 16,400 x g for 10 min at 4 °C. Collect the supernatant and transfer it into a fresh 1.5 mL tube with low protein adsorption. The samples can be stored at -80 °C.
- Determine the protein concentration of the samples with a protein concentration measurement kit according to the manufacturer's protocol (see Table of Materials).
- Separate 200-1000 µg of the same amount of protein in each sample, then add protein lysis buffer + PI to adjust the total volume to 500-1000 µL.
NOTE: The following steps are performed on ice.
4. Preparation of slurry
NOTE: Prepare a 1:1 slurry of protein G gel and epitope tag affinity gel the day before or on the day of immunoprecipitation.
- Preparation of a protein G gel
- Using a tip with the end cut off, mix well and then separate the protein G gel (see Table of Materials) such that 20 µL of beads per sample is prepared.
- Centrifuge at 12,000 x g for 20 s at 4 °C and place the beads on ice for 1 min to allow the beads to level off. Discard the supernatant using a pipette.
- Add 1 mL of protein lysis buffer + PI to the protein G gel prepared in step 4.1.2 and mix by tapping and inverting. Centrifuge at 12,000 x g for 20 s at 4 °C, place the beads on ice for 1 min to allow the beads to level off, and discard the supernatant.
- Repeat step 4.1.3 thrice.
- Add an equal volume of protein lysis buffer to the protein G gel to make a 1:1 protein G gel slurry. The protein G gel slurry can be stored at 4 °C for approximately 1 day.
- Preparation of an epitope tag affinity gel
- Using a tip with the end cut off, agitate well and then separate the epitope tag affinity gel (see Table of Materials) such that 10-15 µL of beads per sample is prepared.
- Centrifuge at 5,000 x g for 30 s at 4 °C and wait for 1 min on ice to allow the beads to level off. Discard the supernatant using a pipette.
- Add 1 mL of protein lysis buffer + PI to the epitope tag affinity gel prepared in step 4.2.2 and mix by tapping and inverting. Centrifuge at 5,000 x g for 30 s at 4 °C, place the beads on ice for 1 min to allow the beads to level off, and discard the supernatant with a pipette.
- Repeat step 4.2.3 twice.
- Add 500 µL of 0.1 M glycine (pH 3.5) to the epitope tag affinity gel prepared in step 4.2.4 and mix by tapping and inverting. This step is performed to remove free epitope tag antibodies. Within 20 min after the addition of glycine, centrifuge at 5,000 x g for 30 s at 4 °C, place the beads on ice for 1 min to allow the beads to level off, and discard the supernatant.
- Add 500 µL of protein lysis buffer + PI to the epitope tag affinity gel prepared in step 4.2.5 and mix by tapping and inverting. Centrifuge at 5,000 x g for 30 s at 4 °C, place the beads on ice for 1 min to allow the beads to level off, and discard the supernatant.
- Repeat step 4.2.6 thrice.
- Add an equal volume of protein lysis buffer to the epitope tag affinity gel to prepare a 1:1 epitope tag affinity gel slurry. The epitope tag affinity gel slurry can be stored at 4 °C for approximately 1 day.
5. Preclearing with the protein G gel and capturing protein complexes with the epitope tag affinity gel
- Mix 1:1 protein G slurry (prepared in step 4) with a pipette fitted with a cut tip and add 40 µL at a time to the sample.
- Rotate the sample for 1 h on a rotator (see Table of Materials) in approximately 30 s per cycle in a refrigerated chamber (4 °C).
- Centrifuge at 12,000 x g for 20 s at 4 °C and place the beads on ice for 1 min to allow the beads to level off.
- Collect the supernatant and transfer it into a new 1.5 mL tube with low protein adsorption.
- Separate the sample for input from the sample in step 5.4 and transfer it into a new 1.5 mL tube.
- Add 20-30 µL of 1:1 epitope tag gel slurry to the sample.
- Rotate on a rotator for approximately 30 s per cycle in a refrigerated chamber (4 °C) for 2 h.
- Add sample buffer to the input prepared in step 5.5 to dilute it to 4x, and heat at 98 °C for 10 min. The input can be stored at -80 °C.
NOTE: Because of the possibility of protein degradation due to freezing and thawing, subsequent steps should be performed without preserving the protein as much as possible.
6. Washing and eluting the precipitated proteins
- Set the temperature of the heat block to 98 °C.
- Centrifuge at 5,000 x g for 30 s at 4 °C and wait for 1 min on ice to allow the beads to level off. Discard the supernatant using a pipette.
- Add 1 mL of protein lysis buffer + PI and mix by tapping and inverting. Rotate on a rotator for approximately 30 s per cycle in a refrigerated chamber (4 °C) for 5 min. Centrifuge at 5,000 x g for 30 s at 4 °C,wait for 1 min on ice to allow the beads to level off, and discard the supernatant.
- Repeat step 6.3 5-10 times.
- Add 10 µL of sample buffer to the sample prepared in step 6.4. Boil at 98 °C for 10 min.
- Centrifuge at 5,000 x g at 4 °C for 30 s.
- Place a column in a new tube with low protein adsorption and transfer the gel to the column using a pipette with a cut tip. A column is used to avoid contamination of the gel.
- Centrifuge at 9,730 x g for 1 min at 4 °C. Collect the flow-through. The sample can be stored at -80 °C.
NOTE: If sample degradation is a concern, the following immunoblotting should be performed without freezing.
7. Immunoblotting
NOTE: Immunoblotting procedures are based on previous reports7,8.
- Load whole samples on a sodium dodecyl-sulfate polyacrylamide gel (SDS-PAGE, see Table of Materials).
NOTE: If necessary, use large-well gels such that the entire sample can be transferred. Gels with 50 µL wells were used here. - Set the gels in the electrophoresis chamber and add 1x Tris/Glycine/SDS buffer (see Table of Materials) up to the appropriate volume around the gels.
- Electrophorese gels at 100 V and 400 mA.
- Transfer onto polyvinylidene fluoride (PVDF, see Table of Materials) membranes.
- Block the PVDF membrane blots with 5% skim milk in PBS-P for 1 h at room temperature.
- Wash the membrane thrice with PBS-P on a shaker at top speed for 5 min. Perform the same procedure thereafter when washing.
- Incubate overnight on a shaker with the primary antibody (see Table of Materials) at 4 °C.
- Wash the membrane thrice with PBS-P the next day.
- Incubate for 1 h with the secondary antibody on a shaker at room temperature.
NOTE: If the molecular weight of the target protein is close to the IgG heavy chain, which has a molecular weight of about 50 kDa, a light chain-specific secondary antibody can be used to avoid overlap of the target band with the IgG heavy chain. This study used light-chain-specific antibodies7 or Veriblot as IP detection reagent (see Table of Materials). - Identify bands of proteins with peroxidase luminescent substrates. The membrane can be saved in PBS after washing it twice with PBS-P.
- Strip for 10 min with a stripping solution (see Table of Materials).
- Wash thrice and block with 5% skim milk in PBS-P. The protocol thereafter is the same as in step 7.6 onwards.
Representative Results
Thermogenic adipocytes, also known as brown and beige adipocytes, have potential anti-obesity and anti-glucose intolerance effects. PR (PRD1-BF1-RIZ1 homologous) domain-containing 16 (PRDM16) is a transcription cofactor that plays an important role in determining thermogenic adipocyte identity9,10.
EHMT1 (euchromatic histone-lysine N-methyltransferase 1), also known as GLP, primarily catalyzes the mono- and dimethylation of lysine 9 of histone H3 (H3K9) using the cofactor S-adenosyl methionine11. A previous report indicated that EHMT1 is an essential methyltransferase in the PRDM16 complex that controls thermogenic fat cell fate through histone methylation12,13. Moreover, we have previously reported that the expression levels of PRDM16 and EHMT1 are significantly correlated with those of thermogenic genes in human perirenal brown adipose tissue (BAT), suggesting that both PRDM16 and EHMT1 play important roles in human BAT development8.
To demonstrate the efficiency of this protocol as a method for confirming PPIs, we transfected DYKDDDDK-PRDM16 and HA-EHMT1 into HEK-293 cells and immunoprecipitated DYKDDDDK-PRDM16 using the above protocol. DYKDDDDK-tagged PRDM16 and HA-tagged EHMT1 were inserted into the cloning sites of the mammalian expression vector pcDNA3.1 (see Table of Materials). Immunoblotting confirmed the association between DYKDDDDK-PRDM16 and HA-EHMT1 in groups transfected with DYKDDDDK-PRDM16 and HA-EHMT1 (Figure 2).
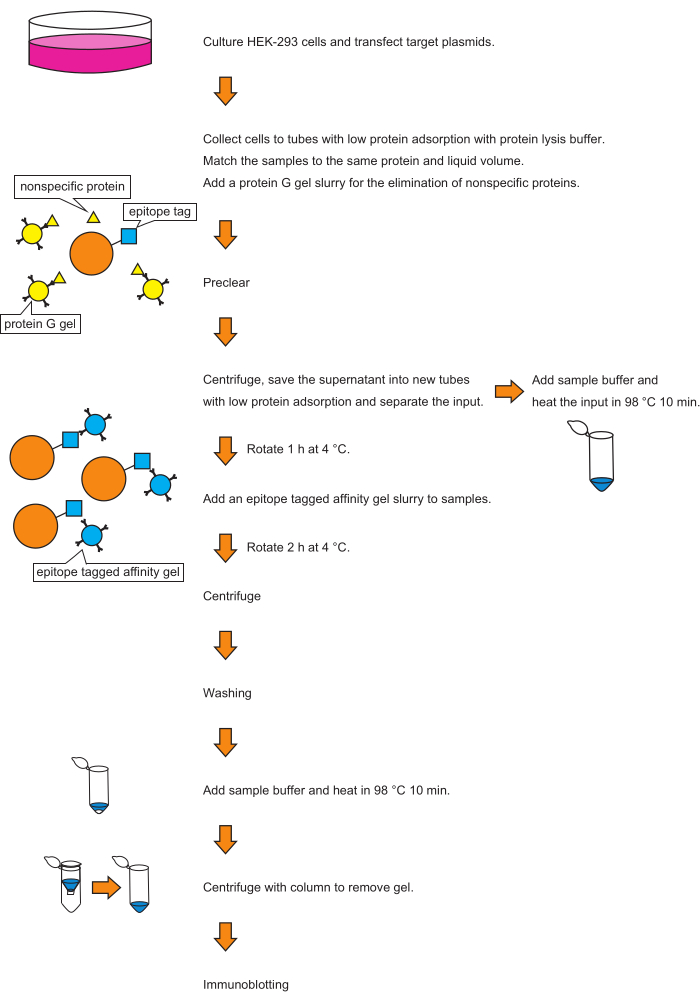
Figure 1: Schematic representation of co-immunoprecipitation. The schematic of the protocol is shown here. The right side shows the workflow of the protocol; the left side shows a graphical representation of the reactions. Please click here to view a larger version of this figure.
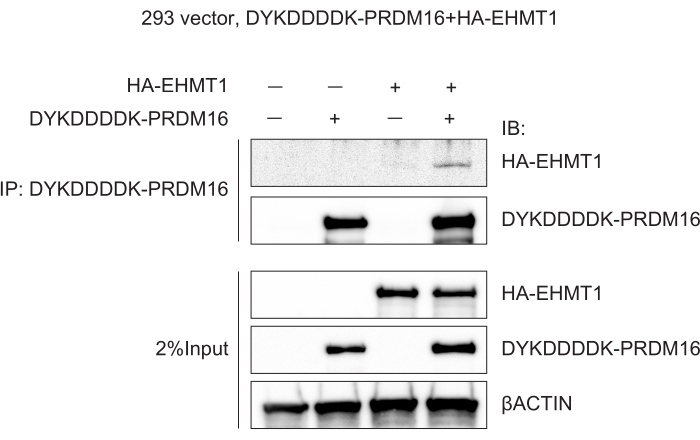
Figure 2: PRDM16 associates with EHMT1. HEK-293 cells were transfected with vector (negative control), DYKDDDDK-PRDM16, or DYKDDDDK-PRDM16 and HA-EHMT1. Co-immunoprecipitation was subsequently performed using a DYKDDDDK tag affinity gel, followed by immunoblotting. Please click here to view a larger version of this figure.
| Protein lysis buffer 250 mL (0.22 μm filtration, store at 4 °C) | ||
| mL | Final concentration | |
| 1 M Tris pH 8.0 | 12.5 | 50 mM |
| 5 M NaCl | 7.5 | 150 mM |
| 0.5 M Ethylenediaminetetraacetic acid | 0.5 | 1 mM |
| Glycerol | 25 | 10% |
| Polyoxyethylene(10) Octylphenyl Ether | 2.5 | 1% |
| DDW | 202 | |
| Total | 250 | |
Table 1: Composition of protein lysis buffer.
Discussion
This protocol is almost like previously reported protocols5,7,14,15. The important point of this protocol is that we never stop the experiment from the cell lysis step to the immunoprecipitation step. Protein degradation hinders PPI detection. Extended time course and repeated freeze-thaw cycles degrade proteins. Electrophoresis in SDS-PAGE should also be performed on the same day of immunoprecipitation not only to minimize protein degradation but also to maximize PPI detection.
In thermogenic adipocytes, the interaction between EHMT1 and PRDM16 was reported in a previous study13. The EHMT1 – PRDM16 complex regulates brown adipocyte cell fate and thermogenesis13. EHMT1 makes PRDM16 stabilize by interrupting the interaction of PRDM16 – Amyloid protein-binding protein (APPBP) 216. This PRDM16 stabilization is regulated by cullin(CUL) 2-APPBP2 and EHMT116.
In this study, HEK-293 cells were used because HEK-293 cells are widely used in the production of recombinant proteins17. Other cell lines that are easy to transfect with plasmids can be used, such as Cos-7 cells and Hela cells. PEI was used for transfection because PEI is more economical and simpler compared to conventional lipofection.
Preclearing, rotation in an epitope tag affinity gel, and washing are particularly important steps in this method. Preclearing can remove the proteins that bind nonspecifically to the gel. In addition, the time for rotation with the epitope tag affinity gel is important because excessive time may result in nonspecific protein detection, and insufficient time may hinder the detection of the target proteins.
The following points should be noted. HEK-293 cells detach easily; therefore, collagen-coated dishes should be used when preparing the samples. PEI solution is cytotoxic; therefore, the medium should be replaced after transfection without leaving the cells for a prolonged period. If PPIs are not observed, the amount of transfected plasmids may be insufficient and/or the amount of protein brought into the IP may be low. In addition, if the number of washes is excessive, the interactions may be missed. To address these, the following can be considered: increasing the amount of plasmid introduced into HEK-293 cells, increasing the amount of protein used for IP, and reducing the number of washes. Furthermore, if too much supernatant is left in the final wash, it will overflow from the wells of the sodium dodecyl sulfate-polyacrylamide gel. If nonspecific bands are observed, the amount of plasmid/protein used for IP may be too high, or the number or intensity of the washes may be insufficient. These can be addressed by reducing the amount of plasmid/protein used for IP, increasing the number of washes, or increasing the washing power by increasing the salt concentration in the washing buffer.
Commonly used detergents such as polyoxyethylene (10) octylphenyl ether may disrupt functionally significant PPIs18. Milder detergents may increase the recovery of functionally important complexes, while they can yield an unacceptable level of nonspecific interactions due to incomplete solubilization. The protein lysis buffer used in the current study contains 1% polyoxyethylene (10) octylphenyl ether. This buffer lyses cytoplasmic proteins with high amounts of proteins overexpressed by the transfected plasmids. When immunoprecipitation is performed on samples prepared by separating nuclei and cytoplasm, the background is diminished compared to samples prepared by whole-cell lysis, and clear target bands can be identified17. If immunoprecipitation is performed with proteins in the nucleus, nuclear proteins can be extracted using a commercial nuclear extraction kit or by destroying and removing cytoplasm with low salt buffer containing detergent and subsequently extracting nuclear proteins with high salt buffer19. The development time in immunoblotting should be adjusted not only to avoid background but also to detect certain bands.
This method had several limitations. First, we used a PPI that is highly expected to detect. Second, because the expression of each gene depends on the combination of plasmids, insufficient protein may be obtained, and the interaction may, therefore, not be detected.
In summary, using this method, PPIs can be confirmed economically and easily compared to mass spectrometry. The PPI between the various epitope tag fused proteins can also be confirmed by using affinity antibodies against each tag instead of the epitope tag affinity gel. Similar immunoprecipitation methods can be performed in other cell types by introducing a plasmid expressing any protein fused to the DYKDDDDK tag. Furthermore, using endogenous antibodies instead of an epitope tag affinity gel makes it possible to confirm endogenous PPIs in various cell types.
Divulgations
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 19K18008 (G.N.), JSPS KAKENHI Grant Number 22K16415 (G.N.), JSPS KAKENHI Grant Number 22K08672 (H.O.), Japan Diabetes Society Research Grant for Young Investigators (G.N.), and MSD Life Science Foundation Research Grant for Young Investigators (G.N.).
Materials
| 0.5 M EDTA (pH8.0) | Nippon gene | 311-90075 | |
| 10% Mini-PROTEAN TGX Precast Protein Gels, 10-well, 50 µL | Biorad | 4561034 | |
| 10x Tris/Glycine/SDS | Biorad | 1610772 | |
| ANTI-FLAG M2 Affinity Gel | Sigma | A2220 | |
| Anti-Mouse IgG, HRP-Linked Whole Ab Sheep | GE Healthcare | NA931-1ML | |
| Anti-Rabbit IgG, HRP-Linked Whole Ab Donkey | GE Healthcare | NA934-1ML | |
| Cell Scraper M | Sumitomo Bakelite | MS-93170 | |
| Collagen I Coat Dish 100 mm | IWAKI | 4020-010 | |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | 4693132001 | |
| DMEM/F-12, GlutaMAX supplement | Invitrogen | 10565042 | |
| D-PBS (-) | FUJIFILM Wako | 045-29795 | |
| Glycerol | FUJIFILM Wako | 072-00626 | |
| Glycine | FUJIFILM Wako | 077-00735 | |
| HA-Tag (C29F4) Rabbit mAb #3724 | Cell Signaling | C29F4 | |
| Laemmli Sample buffer | Bio-Rad Laboratories | 161-0747 | |
| Micro Bio-Spin Chromatography Columns | Biorad | 7326204 | |
| Mini-PROTEAN Tetra Cell for Mini Precast Gels | Biorad | 1658004JA | |
| Monoclonal ANTI-FLAG M2 antibody produced in mouse | Sigma | F3165 | |
| NaCl | FUJIFILM Wako | 191-01665 | |
| pcDNA3.1-FLAG-PRDM16 | This paper | N/A | |
| pcDNA3.1-HA-EHMT1 | This paper | N/A | |
| pcDNA3.1-vector | This paper | N/A | |
| PEI MAX – Transfection Grade Linear Polyethylenimine Hydrochloride | PSI | 24765 | |
| Penicillin-streptomycin solution | FUJIFILM Wako | 168-23191 | |
| Pierce BCA Protein Assay Kit | Thermo scientific | 23227 | |
| Polyoxyethylene(10) Octylphenyl Ether | FUJIFILM Wako | 168-11805 | |
| Polyoxyethylene(20) Sorbitan Monolaurate | FUJIFILM Wako | 167-11515 | |
| Protein G Sepharose 4 Fast Flow Lab Packs | Cytiva | 17061801 | |
| Protein LoBind Tubes | eppendorf | 30108442 | |
| ROTATOR RT-5 | TAITEC | RT-5 | |
| skim milk | Morinaga | 0652842 | |
| Stripping Solution | FUJIFILM Wako | 193-16375 | |
| Trans-Blot Turbo Mini PVDF Transfer Pack | Biorad | 1704156B03 | |
| Trans-Blot Turbo System | Biorad | N/A | |
| Trizma base | Sigma | T1503-1KG | |
| USDA Tested Fetal Bovine Serum (FBS) | HyClone | SH30910.03 | |
| Veriblot | Abcam | ab131366 | |
| β-Actin (13E5) Rabbit mAb #4970 | Cell Signaling | 4970S |
References
- Braun, P., Gingras, A. History of protein-protein interactions: from egg-white to complex networks. Proteomics. 12 (10), 1478-1498 (2012).
- Yanagida, M. Functional proteomics; current achievements. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 771 (1-2), 89-106 (2002).
- Rao, V. S., Srinivas, K., Sujini, G. N., Kumar, G. N. S. Protein-protein interaction detection: methods and analysis. International Journal of Proteomics. 2014, 147648 (2014).
- Hopp, T. P., et al. A short polypeptide marker sequence useful for recombinant protein identification and purification. Nature Biotechnology. 6 (10), 1204-1210 (1988).
- Gerace, E., Moazed, D. Affinity pull-down of proteins using anti-FLAG M2 agarose beads. Methods in Enzymology. 559, 99-110 (2015).
- Einhauer, A., Jungbauer, A. The FLAG peptide, a versatile fusion tag for the purification of recombinant proteins. Journal of biochemical and biophysical methods. 49 (1-3), 455-465 (2001).
- Egusa, G., et al. Selective activation of PPARα maintains thermogenic capacity of beige adipocytes. iScience. 26 (7), 107143 (2023).
- Nagano, G., et al. Activation of classical brown adipocytes in the adult human perirenal depot is highly correlated with PRDM16-EHMT1 complex expression. PLoS One. 10 (3), e0122584 (2015).
- Seale, P., et al. Transcriptional control of brown fat determination by PRDM16. Cell Metabolism. 6 (1), 38-54 (2007).
- Kajimura, S., et al. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes and Development. 22 (10), 1397-1409 (2008).
- Shinkai, Y., Tachibana, M. H3K9 methyltransferase G9a and the related molecule GLP. Genes and Development. 25 (8), 781-788 (2011).
- Able, A. A., Richard, A. J., Stephens, J. M. TNFα effects on adipocytes are influenced by the presence of lysine methyltransferases, G9a (EHMT2) and GLP (EHMT1). Biologie. 12 (5), 674 (2023).
- Ohno, H., Shinoda, K., Ohyama, K., Sharp, L. Z., Kajimura, S. EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature. 504 (7478), 163-167 (2013).
- Bian, W., et al. Protocol for establishing a protein-protein interaction network using tandem affinity purification followed by mass spectrometry in mammalian cells. STAR Protocols. 3 (3), 101569 (2022).
- Cristea, I. M., Chait, B. T. Affinity purification of protein complexes. Cold Spring Harbor Protocols. 2011 (5), (2011).
- Wang, Q., et al. Post-translational control of beige fat biogenesis by PRDM16 stabilization. Nature. 609 (7925), 151-158 (2022).
- Holden, P., Horton, W. Crude subcellular fractionation of cultured mammalian cell lines. BMC Research Notes. 2, 243 (2009).
- Yang, L., Zhang, H., Bruce, J. E. Optimizing the detergent concentration conditions for immunoprecipitation (IP) coupled with LC-MS/MS identification of interacting proteins. Analyst. 134 (4), 755-762 (2009).
- Herrmann, C., Avgousti, D. C., Weitzman, M. D. Differential salt fractionation of nuclei to analyze chromatin-associated proteins from cultured mammalian cells. BIO-PROTOCOL. 7 (6), e2175 (2017).
