1. Plasmid construct
- Amplify the open reading frame (ORF) that codes for the desired IDR by polymerase chain reaction (PCR). Do not include the stop codon since the ORF will be flanked by the genes that encode the fluorescent proteins. For the amplification, design primers with the SacI (5') and BglII (3') restriction sites.
NOTE: For the representative results section, we used AtLEA4-5 as the selected IDR. We amplified the ORF of AtLEA4-5 from the pTrc99A-AtLEA4-5 plasmid12. - Digest the ORF PCR product by consecutive restriction with SacI and BglII enzymes13.
- Obtain the commercial plasmid pDRFLIP38-AtLEA4-5 (#178189) from Addgene (https://www.addgene.org/178189/).
- Conduct a consecutive restriction using SacI and BglII enzymes to remove the AtLEA4-5 ORF from the pDRFLIP38-AtLEA4-5 plasmid, leaving an open plasmid containing the FRET pair13.
- Purify the digested DNA fragments using gel recovery from an agarose gel electrophoresis14. Ligate the restricted fragments using DNA ligase15.
- Transform the cloning reaction into competent Escherichia coli cells (DH5α or related strains) and select in Luria-Bertani (LB) plates containing 50 µg/mL ampicillin16. Grow overnight (ON) at 37 °C.
- Isolate and grow at least five transformed colonies in a new plate and genotype by PCR17. Purify the plasmid DNA from a positive transformed colony using standard miniprep methods18.
- Verify the extraction and integrity of the plasmid DNA using agarose gel electrophoresis19. Verify the correct cloning of the construct using Sanger sequencing20.
2. Plasmid expression in yeast cells
NOTE: Use standard aseptic techniques to perform the following steps. Use a culture laminar flow hood or a lighter.
- Inoculate a streak of S. cerevisiae BJ5465 (ATCC: 208289) strain into 3 mL of Yeast Peptone Dextrose (YPD) medium and grow ON at 30 °C and 200 rpm.
NOTE: BJ5465 is a protease-deficient (CH1) strain recommended for working with IDRs. Other S. cerevisiae strains can be tested and used. - Centrifuge 1 mL of the overnight saturated (OD600 ~ 3-4) yeast culture at 14,000 x g for 1 min. Carefully remove the supernatant by pipetting.
- Resuspend the pellet in 1 mL of Tris-EDTA (TE) buffer (pH 7.5) by gently flicking the tube. Centrifuge at 14,000 x g for 1 min and carefully remove the supernatant.
- Resuspend the pellet in 500 µL of Lazy Bones buffer (40% w/v PEG 3,350; 100 mM lithium acetate; 10 mM Tris-HCl; 1 mM EDTA; pH 7.5) by pipetting. Add 25 µL of boiled (100 °C for 5 min, followed by cooling on ice for 5 min) salmon sperm DNA (2 mg/mL) to the resuspended yeast cells.
- Add 100 ng of the plasmid DNA to the mixture and vortex the mixture for 1 min to ensure thorough mixing. Allow the mixture to stand at room temperature for 1-2 h.
- Heat shock the yeast mixture at 42 °C for 12 min, then cool on ice for another 12 min. Centrifuge the tube at 14 000 x g for 1 min and carefully remove the supernatant.
- Resuspend the pellet in 1 mL of TE buffer (pH 7.5). Plate 100 µL of the transformed cells onto Yeast Synthetic-dropout Medium without uracil (SD-ura) plates supplemented with 15 g/L agar.
- Incubate plates at 30 °C for 2-3 days (Figure 1). Streak at least five yeast transformants in a new plate and grow ON.
3. Validation of yeast transformants
- Pick a small portion of each transformant candidate by gently scraping and resuspending in 5 µL of 20 mM NaOH into individual PCR tubes.
- Heat the sample at 99 °C for 10 min in a thermal cycler to lyse the cells and release the DNA into the solution. This step is crucial for preparing the DNA template for PCR.
- Transfer 1 µL of the boiled sample to a separate PCR reaction mix containing the appropriate primers and PCR reagents for genotyping. We used the following primers: Forward primer: 5´-AAATATACCCCAGCCTCGATCTAGA-3´. Reverse primer: 5´-GTAATACGACTCACTATAGGGCG-3´.
- Set up a PCR reaction and validate the presence of the correct plasmid using agarose gel electrophoresis. Select one transformant for further experiments.
4. Preparation of yeast cell culture for FRET assay
- Inoculate a streak of the selected yeast transformant into 3 mL of liquid 1x SD-Ura medium.
NOTE: Do this procedure in a laminar flow hood and sterile material. - Grow at 30 °C, 200 rpm for at least 12 h to reach saturation. Measure the OD600. Growth should fall between OD600 = 1.0-2.0.
NOTE: If OD600 is lower than 1.0, give cells more incubation time to grow. If OD600 exceeds 2.0, do not proceed and restart from step 4.1.
5. Yeast cell preparation for FRET assay
- Collect 2 mL of the overnight yeast culture and centrifuge at 14,000 x g at room temperature. Carefully remove the supernatant by pipetting.
- Resuspend the pellet in 1 mL of 50 mM 2-(N-Morpholino) ethanesulfonic acid (MES) buffer (pH 6).
- Centrifuge at 14,000 x g at room temperature. Remove the supernatant.
- Repeat steps 5.2 and 5.3. Resuspend yeast cells in 2 mL of MES buffer, pH 6 and pour the volume into a reagent reservoir.
6. Setting up the fluorescence measurements
NOTE: The present scanning is considered to be performed in a microplate reader with fluorescence mode.
- For basic settings, set the instrument as follows: Measurement type: Fluorescence Intensity (FI) spectrum; Microplate name: Greiner 96 F-bottom.
- For optic settings, set the instrument as follows: No. of wavelength scan points: 91; Excitation wavelength: 433 nm; Excitation bandwidth: 10 nm; Emission wavelength: 460 – 550; Step width: 1 nm; Emission bandwidth: 10 nm; Focal height obtained by autofocus; Focal height: 5 mm; Wavelength used for gain: 490 nm.
- For general settings, set the instrument as follows: Settling time: 0.1 s; Reading direction: unidirectional, horizontal left to right, top to bottom.
NOTE: The manually entered gain must be adjusted for every measurement using the well expected to display the highest fluorescence intensity levels throughout the wavelength scan.
7. Preparation of hypertonic solutions and fluorescence measurements
- Prepare solutions of 0 M, 0.2 M, 0.4 M, 0.6 M, 0.8 M, 1 M, 1.5 M, and 2 M NaCl in a 96-well clear-bottom black plate. The final volume in each well will be 200 µL (150 µL of osmolyte solution + 50 µL of yeast cell suspension).
NOTE: Other osmolytes can be used to induce hyperosmotic stress. All the solutions must be prepared in 50 mM MES buffer pH 6. - Load 150 µL of 50 mM MES buffer, pH 6 in first three wells of row A (A1, A2, A3) and in the subsequent wells, add 150 µL of the corresponding solution in increasing order from left to right (Figure 2). To test all the concentrations suggested, continue the loading in row B.
NOTE: This setting considers the measurements of 3 technical replicates for each concentration. - Use a 12-channel micropipette to transfer 50 µL of washed yeast cells to each well. Yeast cells tend to sediment to the bottom of the reservoir. Make sure to resuspend the yeast suspension thoroughly by pipetting up and down before transferring the cells.
- Load cells into the 96-well plate containing the different solutions and mix well by pipetting up and down at least 4x.
- Measure fluorescence intensity emission spectra immediately for the desired wells in a microplate reader with the specification settings of step 6. Repeat the procedure for every IDR to be tested in the FRET assay.
- Optional step. If available, measure a donor-only construct (pDRFLIP38-mCerulean3) following the steps described here.
NOTE: We recommended performing this step to calculate each condition's FRET efficiency. If the reader cannot perform this step, FRET can be calculated using the FRET ratio method. Both methods for FRET calculations are explained in steps 8 and 9. - Perform at least three replicates on three independent days for every construct.
8. Data processing using the FRET efficiency method
NOTE: For readers with knowledge of the R programming language, we provide a set of R scripts to perform data processing described in this section. Scripts can be found at https://github.com/Kaz-bits/cuevaslab-procotols/tree/main/FRET. Follow the instructions in the README file.
- Export data as .xlsx files from the microplate reader.
- Normalize fluorescence intensity values at every scan wavelength to the isosbestic point of the FRET pair used.
NOTE: This step is necessary for comparing the spectra of all conditions tested. The isosbestic point of mCerulean3 and Citrine FRET pair is 515 nm. For example, obtain the ratio of fluorescence intensity values of 460 nm/515 nm, 461 nm/515 nm, 462 nm/515 nm, and so on. - Calculate the mean for every technical replicate once the fluorescence values are normalized. In total, there should be a column for every condition of hyperosmotic stress.
- Visualize all the fluorescence intensity spectra in the same plot (Figure 3). Each spectrum should display a combination of the fluorescence emission spectra of individual mCerulean3 and Citrine. In rare cases, where the FRET efficiency is 0, only the donor fluorescence emission spectrum will be observed, or when FRET efficiency is 1, only the acceptor fluorescence will be observed. The fluorescence emission peak for mCerulean3 (donor) is 475 nm, and the fluorescence emission peak for Citrine (acceptor) is 525 nm.
- Determine if the fluorescence measurement displays a typical FRET change in response to hyperosmotic stress. If the conformation of the IDR is sensitive to the treatment, this will reflect as a change in FRET efficiency. A typical change in FRET efficiency will couple a decrease in the fluorescence intensity of the donor with an increase in the fluorescence intensity of the acceptor or vice versa.
- Quantify the FRET efficiency (EFRET). Obtain the donor (mCerulean3) normalized peak values (475 nm/515 nm) for the IDR construct and the donor-only construct for every condition of hyperosmotic stress. Use the mean normalized peak values of the technical replicates. Perform this operation for each of the three independent replicates. Calculate EFRET with the following formula:
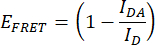
Where IDA is the ratio 475 nm/515 nm from the IDR construct (FRET pair construct), and ID is the ratio 475 nm/515 nm from the donor-only construct. - Compare FRET efficiencies. Normalize the EFRET values of every hyperosmotic stress condition to the EFRET of the non-stress condition (for example, 0 M NaCl). Make comparisons using a box plot or a smooth curve plot. Perform a One-way ANOVA followed by a post hoc Tukey statistical test (p-values: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
9. Data processing using the FRET ratio method
NOTE: If donor-only measurements cannot be acquired, perform the FRET ratio method. This method compares the FRET ratio across the different hyperosmotic stress conditions. Steps 7.1 to 7.5 should be performed before the steps of this section to validate a typical FRET behavior. For readers with knowledge of the R programming language, we provide a set of R scripts to perform data processing described in this section. Scripts can be found at https://github.com/Kaz-bits/cuevaslab-procotols/tree/main/FRET. Follow the instructions in the README file.
- Extract data at 475 nm and 525 nm from the xlsx file previously exported from the microplate reader. These values correspond to the fluorescence emission peak for mCerulean3 (donor, DxDm) and the fluorescence emission peak for Citrine (acceptor, DxAm) when the donor fluorophore is excited (433 nm).
- Normalize fluorescence intensity values at 475 nm and 525 nm to the isosbestic point of the FRET pair used. The isosbestic point of mCerulean3 and Citrine FRET pair is 515 nm. Obtain the FRET ratio (DxAm/DxDm).
- Compare FRET ratios. Normalize the DxAm/DxDm values of every hyperosmotic stress condition to the mean DxAm/DxDm of the non-stress condition (for example, 0 M NaCl). Make comparisons using a box plot or a smooth curve plot (Figure 4 and Figure 5). Perform a One-way ANOVA followed by a post hoc Tukey statistical test (p-values: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
After transforming yeast cells with pDRFLIP38-AtLEA4-5 plasmid, the fluorescence of the positive transformants was observed with a blue light transilluminator and a filter (Figure 1). Preparing the different solutions to induce hyperosmotic stress is time-consuming, so we suggest following the 96-well template of Figure 2. Immediately after the hyperosmotic stress treatment with varying concentrations of sodium chloride, the fluorescence emission spectra were acquired (Figure 3). The fluorescence emission spectra of every condition were obtained to validate a typical FRET change behavior. Normalization of the fluorescence intensity values to the isosbestic point is required to correct experimental errors. For these representative results, we tested the previously reported AtLEA4-5, which is a highly sensitive plant IDR6. As a reference, we also tested the slightly insensitive ABP globular protein (Figure 3). It can be observed that both constructs display a combination of mCerulean3 and Citrine emission spectra (peaks for the donor and acceptor FPs), indicating some degree of basal FRET efficiencies under standard conditions (0 M NaCl). For AtLEA4-5, hyperosmotic stress caused an increase in the fluorescence intensity of the acceptor coupled with a decrease in the fluorescence intensity of the donor, indicating hyperosmotic stress-induced compaction of the IDR (Figure 3A). This trend was not observed in ABP (Figure 3B). To quantify differences in structural sensitivity across the different hyperosmotic stress treatments, we plotted the FRET ratio (DxAm/DxDm) for each condition and performed a One-way ANOVA statistical test (Figure 4). Finally, we compared the structural sensitivity of AtLE4-5 and ABP. We observed that AtLEA4-5 displayed a significantly higher sensitivity than ABP (Figure 5).
Figure 1: Fluorescence emission of S. cerevisiae transformants. SD-ura Petri dish plated with S. cerevisiae transformed with the pDRFLIP38-AtLEA4-5 plasmid. Fluorescence was observed using a blue light transilluminator and an orange filter. Please click here to view a larger version of this figure.
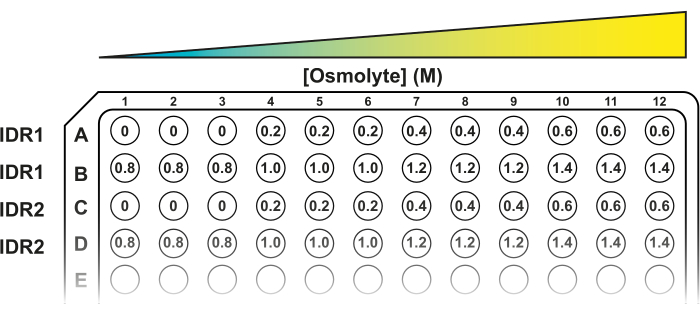
Figure 2: A 96-well plate template for the FRET system. Three technical replicates for each hyperosmotic stress condition are placed in consecutive wells. The first two rows (A and B) fit eight conditions for one construct (IDR1). The following rows can accommodate new constructs (IDR2, IDR3, etc.). Please click here to view a larger version of this figure.
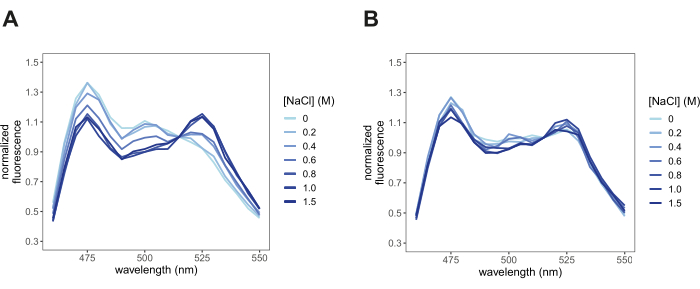
Figure 3: Normalized fluorescence emission spectra under different hyperosmotic stress conditions. Spectra validated a typical FRET behavior in response to hyperosmotic stress (NaCl) treatment. (A) AtLEA4-5, a highly sensitive IDR. The increase in the fluorescence intensity of the acceptor is coupled with the decrease in the fluorescence intensity of the donor. (B) ABP, a slightly insensitive globular protein. Peaks are observed at 475 nm and 525 nm according to the FRET pair mCerulean3 and Citrine. Please click here to view a larger version of this figure.
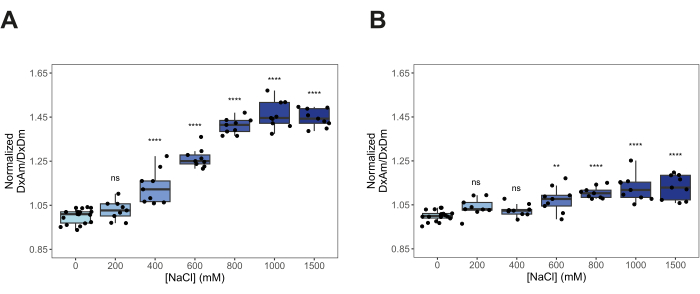
Figure 4: Quantification of structural sensitivity across different hyperosmotic stress conditions. Normalized FRET ratio (DxAm/DxDm) of live yeast cells treated with different concentrations of NaCl. (A) AtLEA4-5. (B) Arabinose-binding protein (ABP). n = 9 independent measurements. One-way ANOVA. ns: non-significant. p-values: *p < 0.05, **p < 0.01, ***p < 0.001, ****p< 0.0001. Boxes represent 25th-75th percentile (line at median). Please click here to view a larger version of this figure.
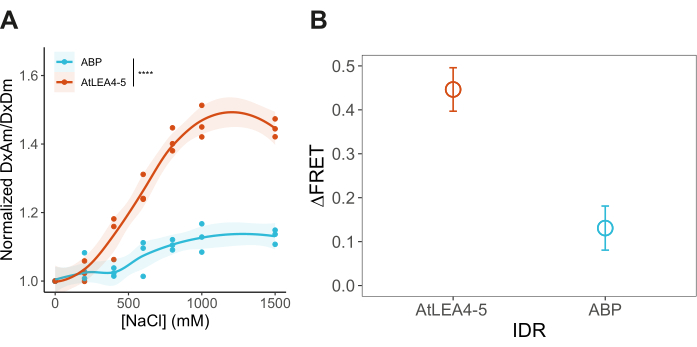
Figure 5: Comparison of the structural sensitivity of AtLEA4-5 and ABP. (A) Normalized FRET ratio (DxAm/DxDm) under different hyperosmotic stress conditions for AtLEA4-5 and ABP. n = 3 independent measurements. (B) Delta FRET value (1.5 M – 0 M NaCl) for AtLEA4-5 and ABP. Change is reported as the structural sensitivity of the constructs measured in the FRET system at 1.5 M NaCl. n = 3 independent measurements. Mean ± SD. Two-way ANOVA using the following p-values: *p < 0.05, **p < 0.01, ***p < 0.001, ****p< 0.0001. Please click here to view a larger version of this figure.
