Monitoring cell morphology throughout the differentiation process
All results described below were generated using the MCND-TENS2 iPSC line for OC differentiation. This iPSC line has previously been used in several studies32,33. Nevertheless, other iPSC lines have also been successfully used with this differentiation protocol.
Regular visual assessment reveals differing and distinct morphological characteristics of iPSCs throughout the differentiation process to OCs (Figure 2). iPSC colonies (Figure 2A) were dissociated into a single-cell suspension, which appears as individual cells throughout the round bottom well plate before centrifugation (Figure 2B). Following centrifugation (step 4.9 in the protocol), cells will collect in the center of the round bottom ultra-low attachment well plate and subsequently form spheres (embryoid bodies, EBs, Figure 2C). EBs will increase two to three times in size throughout the mesodermal differentiation process (Figure 2D) and become easily visible to the eye at the end of the 4-day differentiation period (Figure 2E). EBs can be seen adhering and fusing with the well plate bottom following the transfer into wells of a 6-well plate for hematopoietic differentiation (step 5.3 in the protocol, Figure 2F). After 7-8 days, large quantities of floating hematopoietic cells become visible in the culture medium (Figure 2G). After harvesting and replating hematopoietic cells, an M-CSF maturation phase follows, and OC differentiation is initiated (step 6.4 in the protocol). Within 5-6 days, multinucleated cells with a large transparent cell body first become visible (Figure 2H). A large number of mononuclear cells are still visible at this stage. After 2-3 more days of OC differentiation, OCs will further fuse with adjacent cells to form large polykarions with even more nuclei (Figure 2I).
Assessing the EB-derived hematopoietic population
Hematopoietic differentiation can be performed for a variable length of time. Time periods starting with 7 days up to 9 weeks have been described in the literature. In this protocol, hematopoietic differentiation is performed for a period of 10 days. We found that 10 days of hematopoietic differentiation yielded low numbers of early CD34+ (0.53%, Figure 3A) and a larger number of midterm stage CD43+ (48.5%, Figure 3B) hematopoietic progenitors. More critically, sufficient quantities of CD45+ (96.2%, Figure 3C), CD14+ (33%, Figure 3D), and CD11b+ (35.9%, Figure 3E) HPCs were generated after a treatment period of 10 days to successfully differentiate them further into OCs. However, the cytokine treatment period for hematopoietic differentiation (step 5.4 in the protocol) may need to be adjusted and optimized based on the iPSC line to generate adequate amounts of CD45+, CD14+, and CD11b+ cells.
On average, 6 million hematopoietic cells were harvested after 10 days of differentiation from each well with 8 EBs.
Assessing OC morphology and activity
Following OC differentiation, OCs can be assessed morphologically and functionally. OC precursors are reseeded onto chamber slides or coverslip slides following the M-CSF maturation step to improve image quality when staining for TRAP or Cathepsin K. Enzymatic TRAP staining following OC differentiation shows large, multinucleated, TRAP-positive OCs (Figure 4A). Additionally, slightly TRAP-positive mononuclear cells can be seen interspersed in between multinuclear OCs. Negative controls without the addition of RANKL do not display fused multinuclear OC. Nevertheless, a small number of slightly TRAP-positive mononuclear cells can be depicted (Figure 4B).
Confocal laser scanning microscopy (CLSM) images show OCs stained for Cathepsin K (turquoise) and F-actin (red) in conjunction with DAPI nuclear stain (blue) (Figure 4C, 4D). Large multinuclear OCs are visible when treated with RANKL according to the protocol, which depicts an extensive F-actin cytoskeletal structure and stain positive for Cathepsin K (Figure 4C). Negative controls without the addition of RANKL, on the other hand, do not show fused multinuclear cells.
OCs can be further assessed functionally by measuring the resorptive activity. Bone or mineral resorption assays can be used to determine the resorptive activity. Here, OC precursors were seeded onto calcium phosphate-coated wells and terminally differentiated. Large areas where the mineral coating was resorbed are visible in a bluish-gray color (Figure 4E). Resorption pits of different sizes can be identified. Not resorbed, the remaining calcium phosphate-coating is visible in brown. The presence of resorption pits confirms the identity of the differentiated multinucleated cells as OCs. Additionally, resorption pits can further be quantified in order to assess and compare the resorptive activity. The total area of resorbed mineral over the total well surface area can be quantified as a percentage measure. Additionally, the size and number of resorption pits can further be quantified. Untreated negative controls did not display resorption pits (Figure 4F).
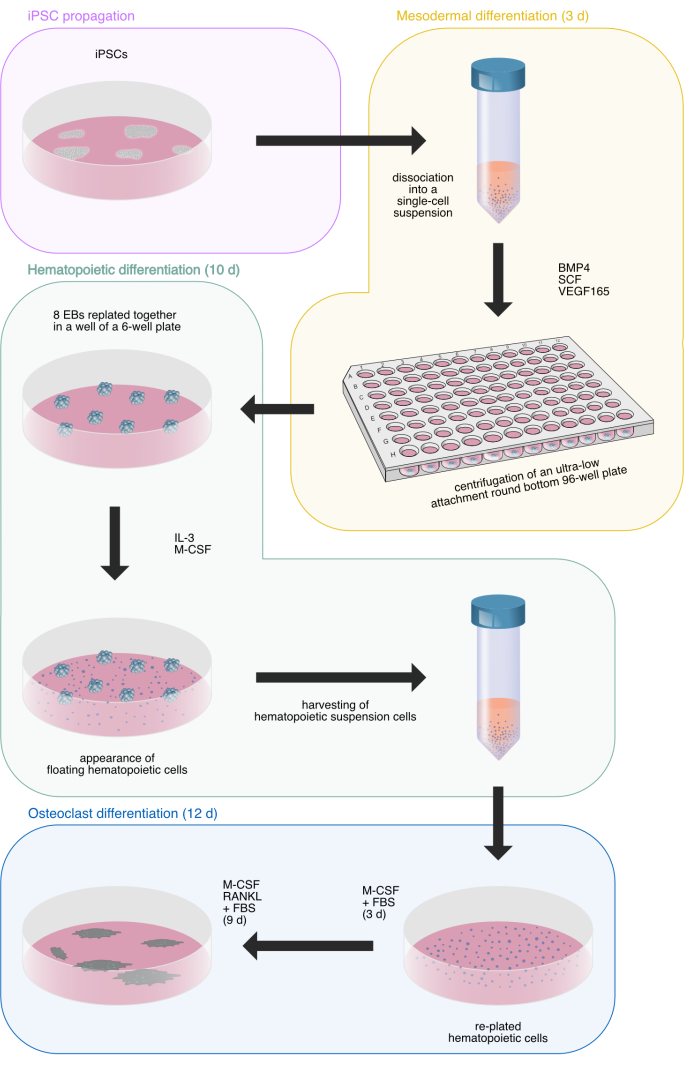
Figure 1: Schematic illustration of the osteoclast differentiation process from human iPSCs. Illustration drawn by Hannah Blümke using Affinity Designer 2.1.1. The illustration utilizes previously used drawings33. Please click here to view a larger version of this figure.
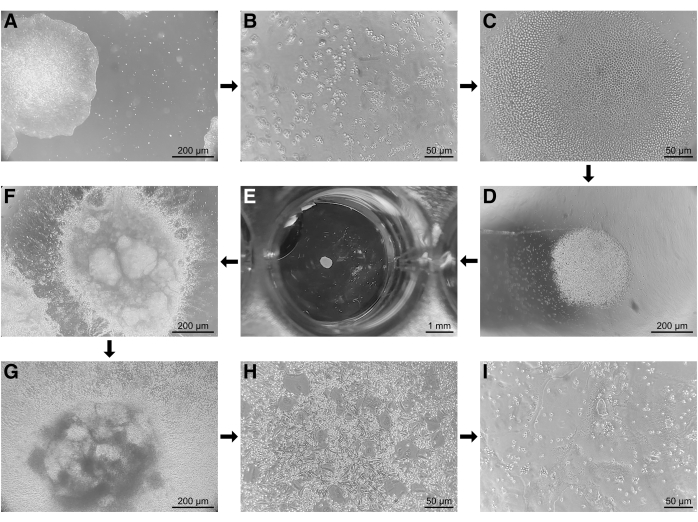
Figure 2: Microscopy images throughout the differentiation process of human iPSCs toward osteoclasts. (A) Undifferentiated iPSC colonies throughout propagation. (B) iPSCs in round bottom wells after dissociation into a single cell suspension prior to centrifugation. (C) Centrally collected single cell iPSCs following centrifugation. (D) EBs grow in size throughout the 4-day mesodermal differentiation period. (E) Visible embryoid bodies following mesodermal differentiation. (F) Following the transfer of embryoid bodies onto basal membrane extract coated 6-well plates, embryoid bodies can be seen adhering and fusing with the well bottom. (G) After 5-7 days of hematopoietic differentiation, a large number of floating hematopoietic cells can be observed in the medium. (H) Following M-CSF maturation, the first osteoclasts with 3-4 nuclei appear after 5-7 days of differentiation with RANKL. (I) At the end of osteoclast differentiation, large multinucleated cells can be observed. Scale bars: A, D, F, G = 200 µm, B, C, H, I = 50 µm, E = 1 mm. Please click here to view a larger version of this figure.
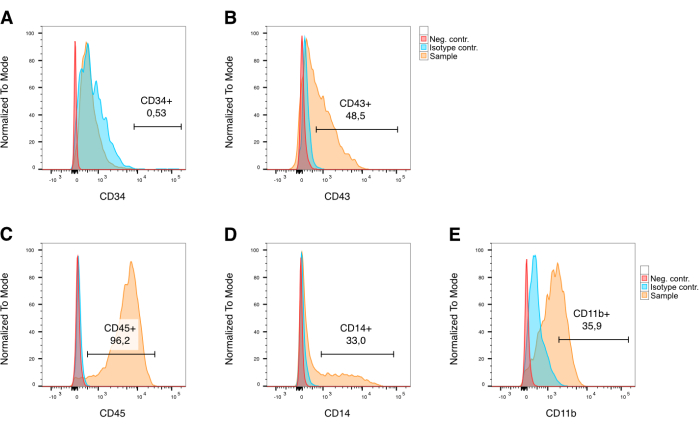
Figure 3: Surface marker analysis of embryoid body-derived hematopoietic cells using flow cytometry. Marker expression allows for the analysis of hematopoietic cells and identification of sub-populations after gating for singlets and live cells. (A) Ontogenetically early CD34+ hematopoietic progenitor cell population is very small to absent in conjunction with this protocol. (B) CD43+ cells make up approximately 50% of the entire population. (C) Later stage CD45+ hematopoietic progenitor cells make up the largest part of the hematopoietic population with 96.2%. (D, E) More direct CD14+ and CD11b+ OC precursors make up 33% and 36%, respectively. In red: unstained negative control, in blue: isotype controls, in yellow: cells stained with the respective marker antibody. Plots use previously published data33. Please click here to view a larger version of this figure.
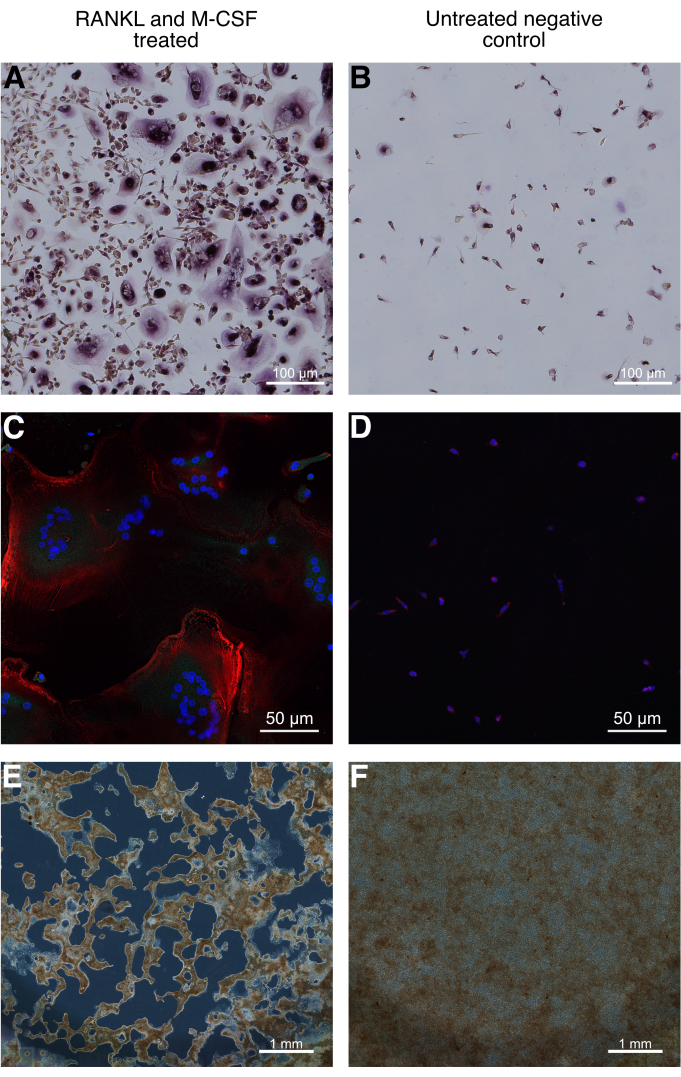
Figure 4: Morphological and functional assessment of iPSC-differentiated human osteoclasts. (A) TRAP staining following osteoclast differentiation shows large, multinucleated, TRAP positive osteoclasts. Slightly TRAP positive mononuclear cells can be seen interspersed in-between multinuclear osteoclasts. (B) Negative controls without the addition of RANKL stained for TRAP do not display fused multinuclear osteoclasts. Nevertheless, a small number of slightly TRAP positive, mononuclear cells can be depicted. (C) Confocal laser scanning microscopy images of osteoclast differentiated hematopoietic cells on a coverslip slide stained for F-actin (red) and Cathepsin K (turquoise) in conjunction with DAPI nuclear stain (blue) show large multinucleated Cathepsin K positive osteoclasts. (D) Negative controls without the addition of RANKL show similar to (B) mononuclear cells at a lower cell density. (E) Functional assessment can be performed by assessing the resorption activity of osteoclasts. For this, osteoclast differentiation is performed on a bone or mineral resorption assay. Tiled well images acquired with an inverted widefield microscope in phase contrast mode depict large resorption areas of the calcium phosphate mineral layer. Resorptive activity can be further quantified by measuring the resorption area over the total area. (F) Negative controls without the addition of RANKL do not show resorption areas. Scale bars: A, B = 100 µm, C, D = 50 µm, E, F = 1 mm. Images have been edited from previously published data33. Please click here to view a larger version of this figure.
