The described methods allow the establishment of SC organotypic slices from mice at stage P3 and their maintenance in culture for a prolonged time in healthy conditions. Moreover, we show a protocol for transplanting cells into the slices and for co-culturing them for up to 30 days (Figure 1). First, we show the optimization of the culture conditions and a protocol suitable for prolonged culturing of the SC slices with transplanted cells (Figure 2A). Slices are generated and maintained from DEV 0 until DEV 2 in the OM, which was originally proposed as an optimal medium for the maintenance of SC slices47. However, due to the presence of serum proteins, this medium could be suboptimal to sustain the neuronal differentiation and maturation of the transplanted neural precursor cells. Indeed, at DEV 3, we tested the switch from OM to the GM, a formulation containing Neurobasal plus B27, which supports neural survival, and without serum, which inhibits the correct neuronal differentiation, promoting instead a glial fate48,49.
Figure 2B shows the results achieved by switching the medium at DEV 3 from OM to GM, compared to the SC slices not receiving the switch (control slices were cultured in OM). We used the distribution of the NFL signal inside the slices as a marker for neuronal integrity (Figure 2B,C). Slices at DEV 7 were healthy in both culturing conditions, showing the diffuse distribution of neurofilament (NFL, in green) inside them. At DEV 10, slices cultured in GM seemed to be healthier with respect to the control slices cultured in OM, as documented by NFL staining distribution. We also estimated the NFL+ area (% NFL+ Area/DAPI+ Area) of the slices shown in the representative images of Figure 2B. The estimated NFL+ area is represented in the histograms in Figure 2C, confirming that the NFL signal is diffusely distributed in the slices at DEV 7 under both conditions. However, at DEV 10, the estimated area covered by NFL staining decreases for the OM culturing condition.
These data suggest that switching to the GM at DEV 3 is well-tolerated for prolonged culturing of SC slices (DEV 10). As a next step, we tested GM at more prolonged time points: DEV 30, DEV 60, and DEV 90. As shown in Figure 3A,B, slices were maintained healthy in culture until DEV 90. NFL staining was found widely present in the slices at each time point, with diffuse sprouting around the slices of neurites departing from the central region. Indeed, we estimated the NFL+ area of the slices shown in Figure 3A and it increased over time as shown in the histograms of Figure 3B. We also observed positivity to the neuronal marker RBFOX3, providing another line of evidence of the neuronal differentiation of the slices. At each time point, we also checked the apoptosis rate by evaluating in different slices the number of cells positive to aCASP3 (Figure 4A,B). The analysis was performed as described in protocol section 10.2. Apoptotic rate (% aCASP3+ cells/total number of DAPI+ cells) was found to be very low at each time point (0.85 ± 0.52%, 0.71 ± 0.27%, 0.66 ± 0.45% for DEV 30, 60, and 90, respectively) with no significant differences between the three considered time points (p-value > 0.05, Figure 4B). These data suggest that the apoptotic rate associated with aCASP3 remains stable during time and, together with the wide distribution of NFL in the slices (Figure 4A), confirm the survival of the slices at each time point.
In support of previous data, we also performed a live/dead assay to evaluate the viability of the slices at the three different time points. We used Calcein (green staining) to label the viable and metabolically active cells and Sytox (cyan staining) to assess cell death. As shown in the histograms in Figure 4C, the percentage of metabolically active cells increases slightly from DEV 30 to DEV 90 (93.17 ± 5.21%, 96.43 ± 3.02%, 96.33 ± 3.10% for DEV 30, 60, and 90, respectively), stabilizing between the last two time points (DEV 30 vs DEV 60 p-value = 0.018; DEV 30 vs DEV 90 p-value = 0.027; DEV 60 vs DEV 90 p-value = 0.99). We found low levels of cell death that decreased over time (6.83 ± 5.21%, 3.57 ± 3.02%, 3.66 ± 3.10% for DEV 30, 60, and 90, respectively) and a significant difference was found between DEV 30 and later time points, DEV 60 and DEV 90 (DEV 30 vs DEV 60 p-value = 0.018; DEV 30 vs DEV 90 p-value = 0.027; DEV 60 vs DEV 90 p-value = 0.99). These data, in association with the apoptosis rate, confirm slice survival over time and support the effectiveness of the long-term culturing protocol performed.
Once the feasibility of prolonged culturing of the SC slices was established, we challenged the system by transplantation of h-SC-NES cells at the first stages of neuronal differentiation. We tested the h-SC-NES cells because they have shown promising results for SCI treatment12. The transplantation procedure of h-SC-NES cells into the mouse SC slices is described in protocol section 6. The SC slices and transplanted h-SC-NES cells were maintained until DPT 30. Cells were grafted at DIV 10 of differentiation (neural precursor stage) into DEV 4 organotypic slices, as shown in the protocol scheme of Figure 5A. Transplanted cells were monitored for the expression of GFP in culture for up to 30 days. Figure 5B shows representative live images, at different DPT, of an SC slice with transplanted GFP+ cells. The stable expression of GFP over time (Figure 5B and Figure 6A) suggests that cells survived into the SC tissue in the previously optimized culture conditions. We also checked the apoptotic rate of transplanted cells as described in protocol section 10.2. The apoptotic rate (% aCASP3+ cells/total number of Hu-Nu+ cells) was found to be very low (0.44 ± 0.34%) after 30 DPT (Figure 6B). Moreover, the apoptotic rate at DPT 30 was found to be in line with that found for the same type of cells at DPT 7, as previously reported40, documenting that the cultures stabilize over time.
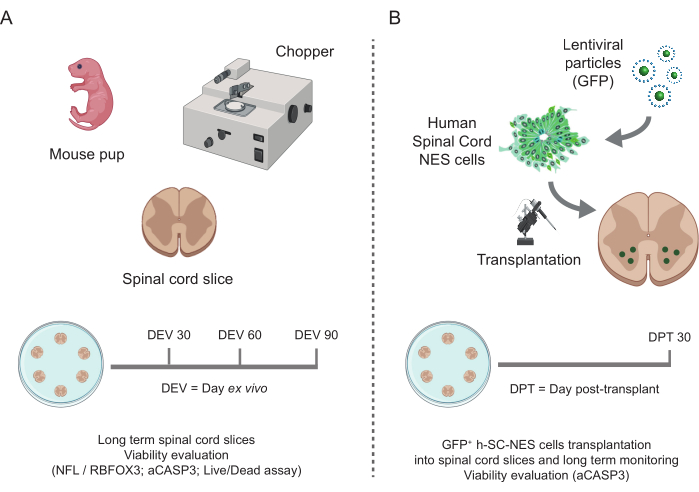
Figure 1: Workflow of the protocol. Representative scheme showing the general workflow of the protocol performed. (A) On the left, a scheme summarizing mouse SC-slice generation from isolated SC of mouse pups at P3 and long-term culturing of SC organotypic slices. (B) On the right, a scheme summarizing the transplantation of h-SC-NES cells expressing GFP into mouse SC-organotypic slices. Grafted cells are maintained for 30 days post-transplant. Abbreviations: h-SC-NES = human spinal cord-derived neuroepithelial stem; GFP = green fluorescent protein; DEV = day ex vivo; DPT = day post-transplant; NFL = neurofilament light chain; RBFOX3= RNA binding fox-1 homolog 3; aCASP3 = active Caspase-3; SC= spinal cord. Please click here to view a larger version of this figure.
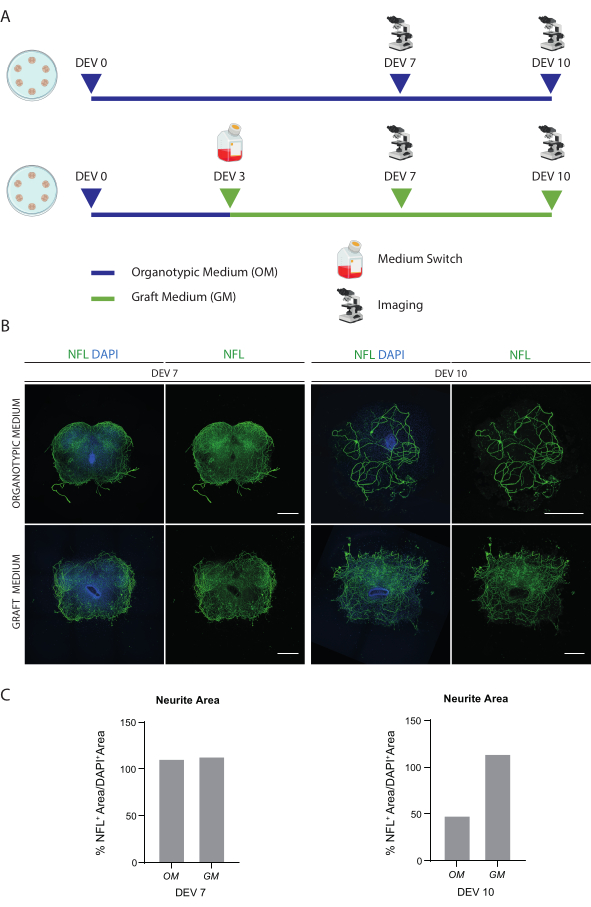
Figure 2: Optimization of long-term culturing conditions. (A) Representative scheme of the protocol for testing OM and GM. OM is maintained until DEV 7-10 for the control group. The medium is switched to the GM at DEV 3 for the treated slices; then, they are fixed at DEV 7-10 for comparison to controls. (B) Representative images comparing mouse SC organotypic slices at DEV 7 and 10 cultured in different conditions. Slices are stained for the cytoskeletal marker neurofilament (NFL, green). The wide distribution of NFL staining in slices cultured with GM suggests an overall survival and differentiation. Nuclei are counterstained with DAPI. Scale bar = 500 µm. (C) Representative histograms of the estimate of the area covered by NFL in the slices shown in Figure 1B. At DEV 10, NFL surface area decreases in the OM culturing condition. Abbreviations: DEV = day ex vivo; DAPI = 4',6-diamidino-2-phenylindole; NFL = neurofilament light chain.; OM = organotypic medium; GM = graft medium; SC = spinal cord. Please click here to view a larger version of this figure.
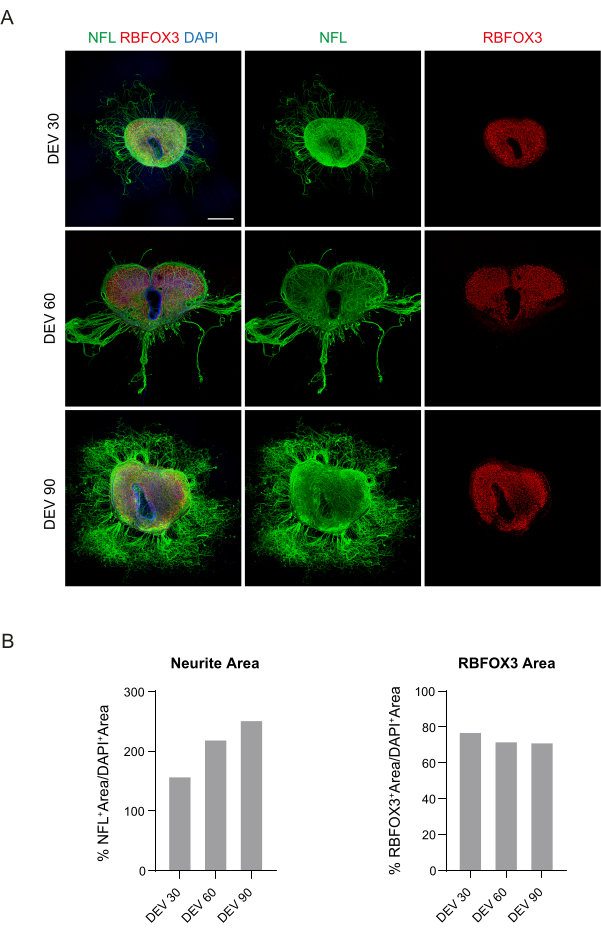
Figure 3: Long-term cultured mouse SC organotypic slices. (A) Slices are maintained in culture until DEV 90. Immunofluorescence assay reveals a wide distribution of the cytoskeletal marker neurofilament (NFL, green) and the nuclear neuronal marker RBFOX3 (red), attesting to their healthy condition and neuronal identity after long-term culturing. Of note, NFL+ axons sprout out diffusely around the slices over time. Nuclei are counterstained with DAPI. Scale bar = 500 µm. (B) Representative histograms of the estimate of NFL+ area and time and, RBFOX3+ area of the slices shown in panel A. NFL+ neurite area increases over time. Abbreviations: DEV = day ex vivo; DAPI = 4',6-diamidino-2-phenylindole; NFL = neurofilament light chain; SC = spinal cord; RBFOX3= RNA binding fox-1 homolog 3. Please click here to view a larger version of this figure.
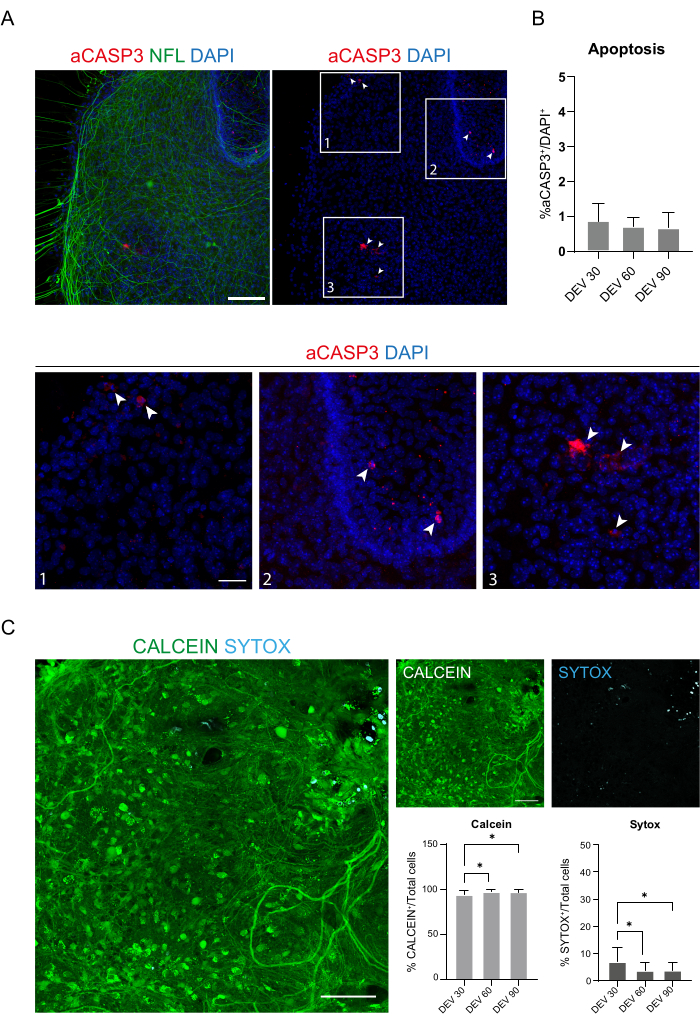
Figure 4: Evaluation of cell viability in the SC slices over time. (A) Representative images of organotypic slices at DEV 60 stained for aCASP3 (red) and NFL (green). Scale Bar = 100 µm. NFL shows a diffuse pattern. Rare cells are positive for the apoptotic marker aCASP3 (Insets: 1-2-3). (B) Analysis of the apoptosis rate in slices at different time points. Mean ± SD, N (replicates) = 6 slices, n (total cells) > 1,000 for each slice, Kruskal-Wallis test, multiple comparison, p-value > 0.05. The apoptotic rate is stable over time. In the insets 1-2-3 of panel A, it is possible to observe details of cells positive for aCASP3 (red staining, white arrows). Small red dots label cell debris and pyknotic nuclei. Scale bar = 50 µm. (C) Representative images of live/dead assay performed on SC slices at DEV 90: metabolically active cells are labeled in green with Calcein, while dead and damaged cells are labeled in light blue (cyan) with Sytox. The two histograms show the % of cells positive for Calcein (on the left) and Sytox (on the right) on the total number of cells. For both mean ± SD, N (replicates) = 6 slices, n (total cells) > 1,000 for each slice, Kruskal-Wallis test, multiple comparison, DEV 30 vs DEV 60 p-value = 0.018; DEV 30 vs DEV 90 p-value = 0.027; DEV 60 vs DEV 90 p-value > 0.99. Abbreviations: DEV = day ex vivo; DAPI = 4',6-diamidino-2-phenylindole; NFL = neurofilament light chain; SC = spinal cord; aCASP3 = active caspase-3. Please click here to view a larger version of this figure.
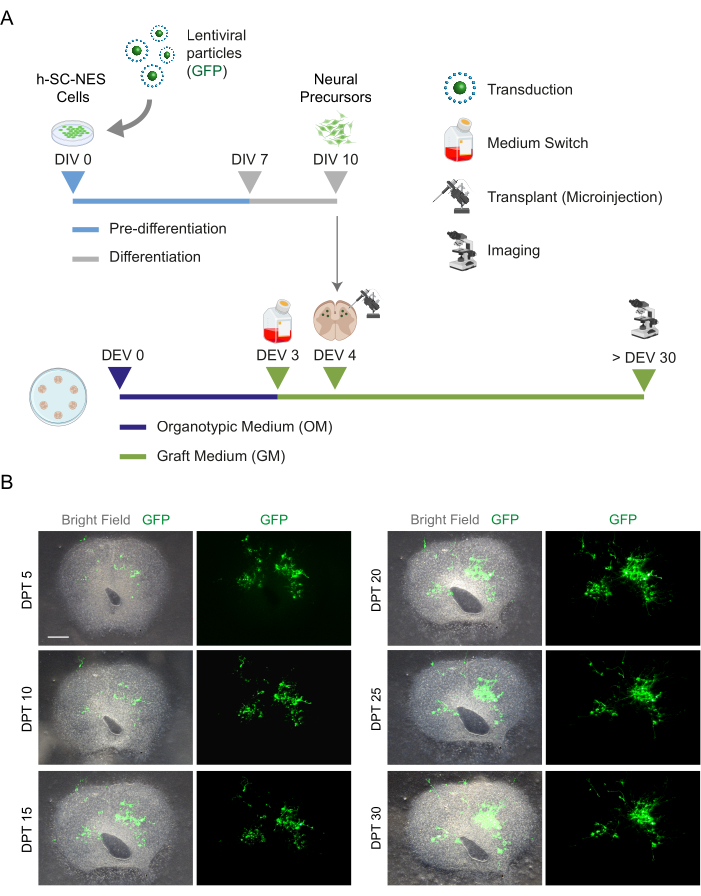
Figure 5: h-SC-NES cell transplantation into mouse organotypic slices. (A) Representative scheme of the transplantation protocol. Cells are transplanted as neural precursors at DIV 10 of differentiation into DEV 4 organotypic slices. (B) Representative images of mouse organotypic slices transplanted with GFP-expressing h-SC-NES cells over time until DPT 30. Cells are transduced with a lentiviral vector carrying the GFP gene. GFP expression over time confirms their viability and adaptation to the slice environment. Scale bar = 500 µm. Abbreviations: DIV = first day in pre-differentiation; h-SC-NES = human spinal cord-derived neuroepithelial stem; GFP = green fluorescent protein; DEV = day ex vivo; OM = organotypic medium; GM = graft medium; DPT = days post-transplant. Please click here to view a larger version of this figure.
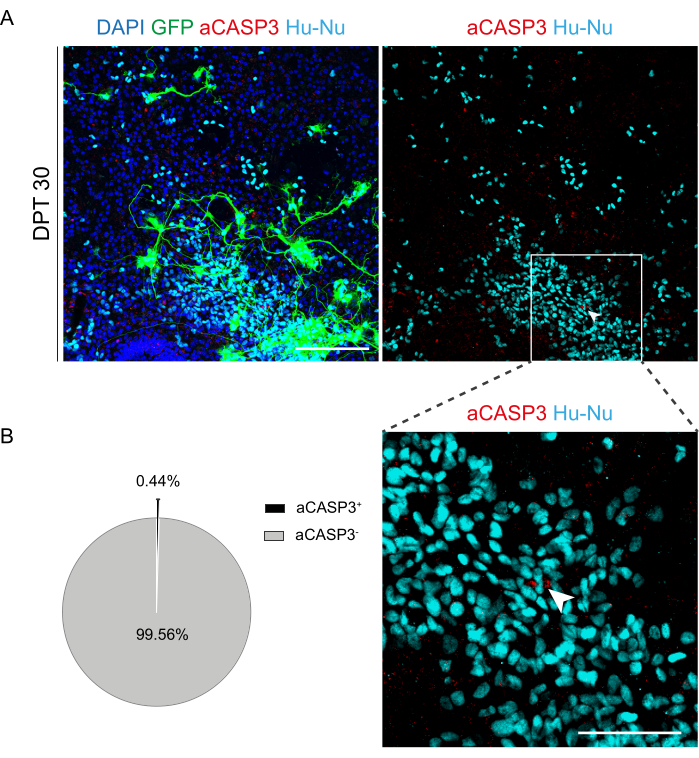
Figure 6: Apoptosis rate evaluation of transplanted h-SC-NES cells after 30 days from transplant. (A) Representative image of a mouse organotypic slice transplanted with GFP-expressing h-SC-NES cells. Cells are transduced with a lentiviral vector carrying the GFP gene for monitoring them into the slices after transplantation. GFP expression over time confirms their viability and adaptation to the slice environment. The time point shown is DPT 30; cells are stained for human nuclei (cyan) and aCASP3 (red). Scale Bar = 150 µm. (B) On the left, representative pie chart of the apoptosis analysis of cells transplanted in slices at DPT 30 (N (replicates) = 5 slices, n (cells) = 5,000), and on the right, an inset of Hu-Nu+ cells and a detail of a cell positive to aCASP3 (white arrow). Scale bar = 75 µm. Small red dots label cell debris and pyknotic nuclei. Abbreviations: h-SC-NES = human spinal cord-derived neuroepithelial stem; GFP = green fluorescent protein; DPT = day post-transplant; DAPI = 4',6-diamidino-2-phenylindole; NFL = neurofilament light chain; aCASP3 = active caspase-3; Hu-Nu = human nuclei. Please click here to view a larger version of this figure.
Table 1: Composition of solutions used in this protocol. Please click here to download this Table.
