Preliminary note
Surgery must be performed by fully trained investigators upon approval from the local authorities for animal welfare. All precautions must be taken to minimize the pain and stress inflicted to the animal including deep anesthesia, administration of postoperative analgesia and, for operations lasting more than 10 min, application of an eye lubricant to prevent corneal dehydration and blindness. Anesthetized mice must be kept constantly warm at 37 °C until full recovery and every surgical tool and solution must be sterile. Although the use of a biological hood is not strictly necessary, the operator must take all reasonable precautions to minimize the possibility of infecting the animal during the operation by wearing a lab coat, mask, and gloves. Similarly, every step concerning the generation and use of viruses must be authorized and must be performed in S2 areas according to the regulations of the local authorities. Finally, a thorough decontamination of all tools and surfaces that could have been in contact with viruses must be performed according to approved facility disinfection practices (for HIV by wiping with 70% Et-OH and/or autoclaving).
Part 1: Expansion of Embryonic NSC
1. In utero electroporation
- After cloning the transgenes (i.e. cdk4/cyclinD1) in pCMS-EGFP (Clontech) or pDSV-mRFPnls vectors 16, purify the plasmids using EndoFree kit and resuspend in sterile PBS to a concentration of 3-5 μg/μl. Soon before surgery, mix the plasmids with 0.05% FastGreen in PBS at a final ratio of ca. 4:4:1 and centrifuge the mixture for 2 min at 16,000 x g to remove all precipitates.
- Load the DNA mixture into a previously pulled borosilicate glass capillary. Pulling parameters using a P-97 pipette puller are: pull: 200; vel: 140; time: 140. Heat is given by a ramp test and depends on the specific lot of capillaries being used. Mount the capillary on the nozzle of the PicoPump that is near to the in utero electroporation platform (Figure 1A) and, under a stereomicroscope, bend its tip and nick it at the inflection point.
- Sterilize all surgery tools in a dry glass bead sterilizer and deeply anesthetize a pregnant mouse at day 12-15 of gestation using an isoflurane vaporizer. Place the animal in supine position on a heating platform set at 37 °C and keep under constant isoflurane administration through a nose cone.
- Shave the skin of the abdomen, disinfect with Betaisodona multiple times, eventually wiping in between with 70% ethanol, and inject buprenorphine (diluted in PBS) subcutaneously at 0.1 mg/kg concentration as pre-emptive analgesic. Using fine scissors, make a 2 cm longitudinal cut of the skin and, subsequently, of the underlying muscular wall to access the peritoneal cavity. Dispense in the peritoneal cavity ca. 2 ml of IUE solution (D-PBS containing 100 U/ml of pen/strep) prewarmed at 37 °C and keep the solution on a heating block.
- Cover the mouse with sterile drap containing a fissure from which the uteri will be removed. Retract the incision using a tungsten retractor, identify the uterus and pull it out holding it with forceps between adjacent embryos (Figure 1B; left) and finally lay it down on the sterile drap. During the whole operation, rinse the uterus with IUE solution to moisturize the organ and body cavity and prevent dehydration of the animal.
- Handle the uterus carefully holding it between thumb and index and turn one embryo until its head is visible and oriented towards the operator. Identify the telencephalic hemispheres and inject one of them from the dorso-lateral side. Release 1-2 μl of the DNA solution using the footswitch of a PicoPump until the ventricle is outlined by FastGreen (Figure 1B; right).
- Place the anode of the electrodes on the injection site and the cathode on the contralateral side (Figure 1C) and deliver 6 pulses of 30 V for 50 ms with 950 ms interval between each pulse using an electroporator generating square-shaped electric fields operated through a footswitch. Avoid placing the electrodes close to the placenta as this may cause hemorrhage and damage the embryo.
- When all embryos are injected and electroporated, place the uterus back into the abdominal cavity and close the muscular walls with a suture string suturing every 3-4 mm. The knots should be snug to the muscle but not too tight to allow a little swelling of the tissue. Finally, close the overlying skin using metal clips. Disinfect the skin with Betaisodona, remove the mouse from the isoflurane mask and transfer it in a housing cage when fully awake. Monitor the recovery of the mouse until locomotor behaviour is normal.
2. Processing of the sample
- One or more days after electroporation, sacrifice the mouse, collect the embryos and identify those correctly electroporated using a fluorescent stereomicroscope (Figure 1D). Dissect the brains on ice-cold PBS, and fix them overnight in 4% PFA (paraformaldehyde in phosphate buffer pH 7.4). Eventually, BrdU can be administered before sacrifice according to different paradigms to investigate cell cycle kinetics and cell proliferation 3.
- The brains are then processed for sectioning and immunostaining for the several markers of radial glia, basal progenitors and neurons (Pax6, Nestin, Tbr2, Tbr1, Tuj1 etc.) to evaluate the effect of the functional transgenes on targeted NSC and their progeny that are identified by the expression of the co-electroporated reporter gene.
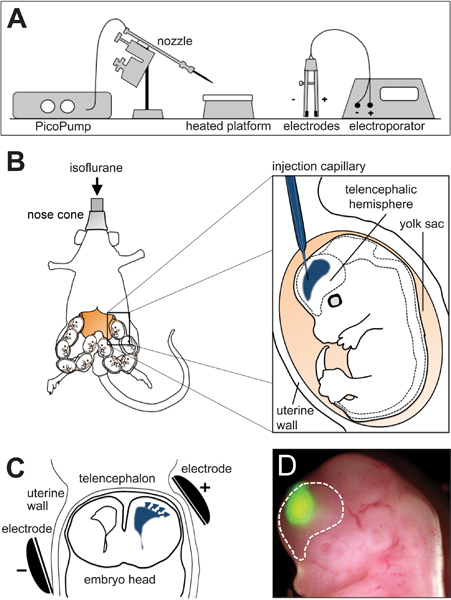
Figure 1. In utero electroporation. (A) Schematic representation of an in utero electroporation platform. (B) Drawings showing the positioning of the pregnant mouse during surgery (right) and injection of the DNA/FastGreen mixture (blue) in the telencephalic hemisphere of a mouse embryo (left). (C) Scheme showing the placement of the electrodes in contact with the uterine walls with the anode (+) adjacent to the site of injection (blue). Arrows indicate the migration of the DNA towards the anode. (D) Picture of a mouse embryo 24 hr after electroporation with GFP plasmids at embryonic day 13. Dashed line demarcates the telencephalic hemisphere. Scheme in A modified from Calegari et al., 2004 17.
Part 2: Expansion of Adult NSC
1. Production of HIV-derived viral particles
- Day 1: plate 5 x 106 293T cells on a 10 cm dish with 8 ml of D-MEM containing 10% of heat inactivated fetal bovine serum and 100 U/ml of pen/strep (culture medium). Use 15 dishes for one viral preparation and culture overnight at 37 °C, 5% CO2.
- Day 2: for every dish, dilute in 1 ml of plain D-MEM 5 μg of each of 3 plasmids encoding for i) VSVG (vesicular stomatitis virus type G) envelope protein, ii) gag/pol (group specific antigen/polymerase), and iii) a polycistronic construct expressing the functional and reporter transgenes (i.e. cdk4/cyclinD1/GFP) previously cloned in a HIV-based transfer vector (p6NST90)9. Mix this solution with 1 ml of D-MEM containing 45 μl of polyethylenimine that has been dissolved beforehand in PBS at 1 mg/ml stock solution, and incubate for 15-30 min at room temperature. In the meanwhile, remove the media from the cells and add 4 ml per dish of fresh D-MEM containing 15% of heat inactivated fetal bovine serum and 100 U/ml of pen/strep. Add the 2 ml of transfection mixture and culture overnight.
- Day 3: add 120 μl of 500 mM Na-butyrate per dish to enhance the expression of the transgenes, mix gently and culture for 6-8 hr after which replace the transfection medium with 5 ml of culture medium.
- Day 4: collect the supernatants (ca. 70 ml total), pass through a 0.22 μm filter and transfer in 2 polyallomer centrifuge tubes (35 x 89 mm). Ultracentrifuge at 110,000 x g for 90 min at 4 °C using a swing rotor. Discard the supernatant, add 6 ml of PBS to each tube and incubate on ice for 1 hr. Resuspend the pellet and transfer it in one polyallomer centrifuge tube (14 x 95 mm). Ultracentrifuge at 110,000 x g for 90 min at 4 °C using a swing rotor. Discard the supernatant, resuspend the pellet in 50 μl of PBS, make 5 μl aliquots and store at -80 °C. Viral titer will be in the range of 108-109 cfu/ml. (Note: viruses are very sensitive to temperature and storage, avoid re-freezing, keep on dry ice during transport and thaw soon before use.)
2. Stereotaxic injection
- Anesthetize a 6-10 weeks old mouse with isoflurane and place the animal in prone position on a stereotaxic frame (Figure 2A) using the ear bars and the palate support to properly fix the head. The animal is kept warm on a heating pad set at 37 °C and under constant isoflurane administration.
- Inject subcutaneously 100 μl of buprenorphine working solution as pre-emptive analgesic, shave a stripe of skin on the head of the mouse, and desinfect as described in 1.4. Using a scalpel, make a ca. 1.5 cm long longitudinal incision on the head skin exposing the skull at the level of the sagittal suture. Retract the skin and gently clean the bone surface with a sterile cotton bud.
- Half-fill a glass capillary (specifications in 1.2) with paraffin oil and mount it on the injector pump inserting the capillary until the second ring (Figure 2B). Move the capillary holder in the x, y and z axis until the tip of the capillary is positioned on the bregma, the conjunction point between the sagittal and lateral sutures (Figure 2C) (to be determined with the help of a stereomicroscope), and re-set x, y, and z values to 0.
- Move the capillary by ±1.6 mm in the medio-lateral axis (x) and -1.9 mm in the anterior-posterior axis (y) and mark the bone using a surgical marker pen. Make a hole of ca. 0.5 mm diameter on the skull using an electric driller paying attention not to damage the brain.
- Put a 5 μl droplet of viral suspension (1 aliquot) on a piece of parafilm placed on the ear bars and immerge the tip of the capillary in the droplet. Suck ca. 3.5 μl of viral suspension using a nanoliter injector set at 55 nl/sec. Execute this step rapidly as the droplet will evaporate.
- Move the capillary to the site of injection and move it down until the tip touches the pial surface. Reset the dorso-ventral axis value (z) to 0. At this point, penetrate the tissue with the capillary for -1.9 mm and release 1.5 μl of viral suspension at a rate of 4 nl/sec. Wait for 2-3 minutes to minimize the backflow of viral suspension trough the capillary track and then retract the capillary. A second injection can be performed in the controlateral hemisphere.
- Suture the skin, disinfect, and allow the animal to recover from anesthesia as described previously for in utero electroporation (1.8).
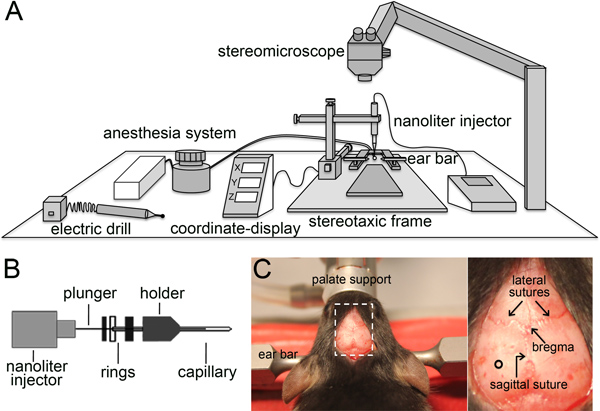
Figure 2. Stereotaxic viral injection. (A and B) Schematic representation of the components needed to perform stereotaxic injection (A) and a disassembled capillary holder showing the insertion of a capillary until the second ring (B). (C) Picture of the mouse head immobilized by the ear bars and palate support with the skin cut to expose the skull (left). The magnification of the dotted box (right) shows the lateral and sagittal sutures, the bregma, and the site of injection (circle).
3. Processing of the sample
- One-to-three weeks post-infection, perfuse the mouse with ca. 100 ml of 4% PFA, dissect the brain and post-fix it overnight in 4% PFA. Eventually, BrdU and/or tamoxifen can be administered before sacrifice according to different paradigms to investigate cell cycle kinetics and to stop the expression of the viral transgenes flanked by loxP sites, respectively 9.
- The brain can then be processed for immunostaining using several markers of stem and progenitors cells and neurons (e.g. Sox2, GFAP, Nestin, Tbr2, doublecortin, etc).
- Overall, in utero electroporation yields 60-80% of electroporated embryos displaying strong expression of the reporter gene in a wide area of the lateral cortex (Figure 3A). Fluorescent proteins can be easily detected on whole mount embryos as soon as 3-6 hours after surgery with the signal lasting for more than one week. Within the targeted area, about 30-50% of cells usually express the transgene(s) with the proportion of cells co-expressing two (or more) co-electroporated transgenes being usually above 90% (Figure 3B, left) 6,8,15. While a minimal level of apoptosis (ca. 2.0-2.5%) could be observed after about 2 hours from electroporation, this value will return to physiological levels (ca. 0.5-1.0%) after about 6 hours (FC, unpublished data). The proportion of embryos or pregnant mice dying due to the surgery is typically negligible (<5%). Co-electroporation with cdk4/cyclinD1 under these conditions followed by 48 hours of development triggers an increase in the expansion of neural progenitor by 30% (Figure 3B, right, and C) 15.
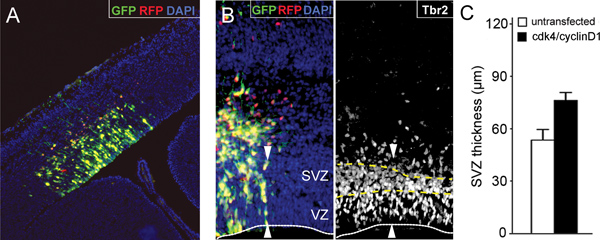
Figure 3. Expansion of neural progenitors by cdk4/cyclinD1 overexpression. (A) Fluorescence picture of a section through the lateral cortex of a mouse embryo 24 hours after electroporation with GFP and RFP plasmids at embryonic day 13. (B) Fluorescent pictures as in A 48 hours after co-electroporation with cdk4/GFP and cyclinD1/RFP plasmids and immunolabeling for the basal progenitor marker Tbr2. Fluorescent reporters with DAPI counterstainig (left) and Tbr2 (right) are shown. Lines indicates the apical surface of the ventricular zone (VZ; white line) and the boundaries of the subventricular zone (SVZ; yellow dashed lines). Note the increased thickness of the SVZ in the electroporated portion of the cortex (white arrowheads) due to an increased generation and expansion of basal progenitors after overexpression of cdk4/cyclinD1. (C) Quantification of the effect shown in B. Bars=SD; p<0.05; n=3. Bar graph in C is taken from Lange et al., 2009 15.
- After viral stereotaxic injection, ca. 80% of operated mice display strong expression of the transgenes throughout the whole dentate gyrus. Constitutive expression of GFP can be easily detected on 40 μm sections as soon as 4 days after surgery. Along the rostral-to-caudal axis of the dentate gyrus, about 10-30% of cells (equivalent to a total of ca. 10,000-30,000 cells) usually express the transgene(s) (Figure 4A) 9. The proportion of mice dying due to the surgery is negligible (<5%). Three week overexpression of cdk4/cyclinD1 under these conditions triggers an increase in NSC by over two-folds (Figure 4B) while decreasing neurogenesis by 70% (Figure 4C) 9.
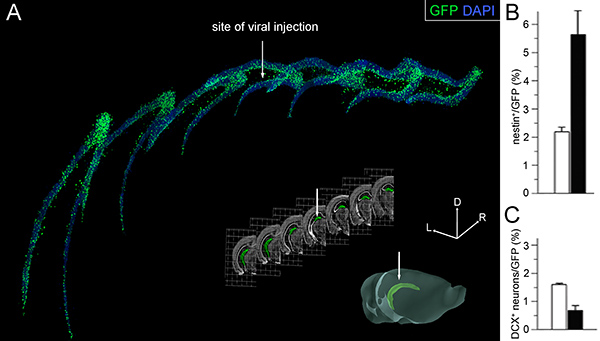
Figure 4. Effects of viral infection. (A) 3D reconstruction of 7 fluorescence pictures taken from 40 μm thick serial coronal sections (collecting 1 every 6) along the rostral-to-caudal axis of the hippocampus. GFP+ infected cells (green) and DAPI nuclear staining (blue) of the dentate gyrus are shown (top). Bright field pictures of the coronal sections corresponding to those shown in A (middle) and 3D representation (bottom) of the whole cerebral hemisphere with the hippocampus highlighted in green pseudocolor (modified from the Allen Brain Atlas; www.brain-map.org). White arrows point the site of injection. Lateral (L), rostral (R), and dorsal (D) axes (x, y, and z stereotaxic coordinates) are indicated (right). (B and C) Proportion of nestin+ NSC (B) and DCX+ newborn neurons (C) within the population of GFP+ infected cells 3 weeks after stereotaxic injection of GFP (white) or cdk4/cyclinD1/GFP (black) viruses. Bars=SD; p<0.005; n=3. Graphs in B and C taken from Artegiani et al., 2011.
