Visualization and Analysis of mRNA Molecules Using Fluorescence In Situ Hybridization in Saccharomyces cerevisiae
Summary
This protocol describes an experimental procedure for performing Fluorescence in situ Hybridization (FISH) for counting mRNAs in single cells at single-molecule resolution.
Abstract
The Fluorescence in situ Hybridization (FISH) method allows one to detect nucleic acids in the native cellular environment. Here we provide a protocol for using FISH to quantify the number of mRNAs in single yeast cells. Cells can be grown in any condition of interest and then fixed and made permeable. Subsequently, multiple single-stranded deoxyoligonucleotides conjugated to fluorescent dyes are used to label and visualize mRNAs. Diffraction-limited fluorescence from single mRNA molecules is quantified using a spot-detection algorithm to identify and count the number of mRNAs per cell. While the more standard quantification methods of northern blots, RT-PCR and gene expression microarrays provide information on average mRNAs in the bulk population, FISH facilitates both the counting and localization of these mRNAs in single cells at single-molecule resolution.
Introduction
Using bulk measurement techniques, it is not possible to assay the number of transcripts or transcriptional activity within single cells1. Using fluorescent proteins driven by promoters of interest as reporters of gene expression can address this issue to some extent, but the time required for fluorescent proteins to fold obscures early dynamics. Long-lived fluorescent proteins also cannot report mRNA lifetimes. The FISH method can be used to assay mRNA during its complete life cycle, from transcription initiation in the nucleus to subsequent maturation and decay in single cells, with single-molecule resolution.
The original in situ experiments for visualizing nucleic acids used radiolabeled RNA probes to probe DNA elements. These included visualizing ribosomal DNA in ovaries of the frog Xenopus laevis 2 and satellite DNA in mouse tissue3. The first fluorescent in situ experiment used an RNA molecule marked with a fluorophore to probe particular DNA sequences4. The first application of fluorescent probes for visualizing RNA in situ was the visualization of actin gene expression in chicken muscle tissue culture5. More recently, in budding yeast, FISH has been used to investigate oscillations in transcription during the yeast metabolic cycle6, the decay of mRNAs during cell cycle progression7, and spatial localization of mRNA transcripts during mitosis8. FISH has been used in yeast to show that uncorrelated fluctuations in constitutively transcribed genes, which constitute more than half of all yeast genes, arise from uncorrelated transcription initiation9. In non-yeast species, FISH has been used to identify stem-cell markers in the mouse intestine10 and to determine that incomplete penetrance of cell fates can result from stochastic gene expression fluctuations in C. elegans embryos11.
The FISH method described here works by hybridizing dye-labeled, single-stranded DNA probes to mRNA messages. Cells are imaged and mRNAs are counted using a spot-detecting algorithm. Single-stranded probes can be generated with a DNA synthesizer and then labeled (referred to here as Singer probes) or ordered commercially as pre-labeled probes (Stellaris probes)12,13. A major difference between the Singer and Stellaris probes is that the Singer probes are longer (~50 bp) and are multi-labeled while the Stellaris probes are short (~20 bp) with only one label per probe, as described by Raj et al 14. Additionally, the Stellaris approach uses many more probes per gene than that of Singer (~30 versus 5 probes per gene, respectively). Below we provide a protocol that describes the use of either type of probe. In section 2, we provide a protocol for labeling amino-allyl thymidine-containing probes with a chosen Cy dye. An overview of the computational steps required to identify single mRNA spots is provided in Section 7.
Protocol
Figures 1 and 2 are schematics of the FISH experimental procedures and image analysis pipeline used for quantifying FISH images.
1. Solutions to Prepare
*The solutions below are for use with Singer probes. If using Stellaris probes, replace “40% formamide” with “10% formamide” in both Hybridization Buffer and Wash Buffer. Additional changes to Hybridization Buffer when using Stellaris probes are (1) add 1 g Dextran Sulfate and (2) do not include 10 mg ssDNA.
Buffer B
8 ml 1 M KH2PO4
41.5 ml 1M K2HPO4
109.3 g Sorbitol
Spheroplasting Buffer
890 μl Buffer B
100 μl VRC
10 μl 25,000 U/ml Lyticase
2 μl β-mercaptoethanol
Hybridization Buffer (10 ml, final volume)
10 mg E. coli tRNA
10 mg ssDNA*
100 μl 200 mM VRC stock
40 μl of 50 mg/ml BSA
1 ml 20X SSC
4 ml 40% Formamide*
Nuclease Free Water (to 10 ml final volume)
1 g Dextran Sulfate*
*Hybridization Buffer may be kept in 0.5 ml aliquots at -20 °C for convenience.
Wash Buffer (50 ml, final volume)
5 ml 20X SSC
20 ml 40% Formamide*
Nuclease Free Water (to 50 ml final volume)
Labeling Buffer
1.06 g Sodium Carbonate
100 ml DEPC Water
pH 9
2. Probe-labeling (Singer Probes Only)
We obtain these probes by in-house synthesis using an ABI oligonucleotide synthesis apparatus. Typically, 4-5 ~50 bp oligonucleotides are synthesized that are homologous to the gene of interest, substituting amino-allyl thymidine for several thymidines spaced at least 8, preferably 10+ bp apart. Because of their sensitivity to ozone, we work in an ozone-free facility when using CY dyes.
- Obtain ~5 probes and resuspend in 100 μl water – check concentrations on Nanodrop.
- Depending on how many probes/gene, combine total of 10 μg oligonucleotides/gene (e.g. if have 5 probes/gene, then want 2 μg/probe).
- Use QIAquick columns to purify probes according to QIAquick Nucleotide Removal Kit Protocol.
- Add 10 volumes Buffer PN to total volume of combined probes and mix.
- Apply sample to QIAquick column – if total volume is greater than 750 μl, spin down twice using half of volume in each spin
- Let stand for 1 min.
- Centrifuge 1 min at 6,000 rpm.
- Wash with 750 μl Buffer PE.
- Centrifuge 1 min at 6,000 rpm.
- Dispose of flow through and re-spin column for 1 min at 13,000 rpm to dry.
- Place QIAquick column in new microcentrifuge tube and elute DNA with 50 μl H2O – Make sure H2O pH is within 7.0 and 8.5 and is placed directly on membrane.
- Let stand for 1 min.
- Centrifuge for 1 min at 13,000 rpm to elute DNA.
- Lyophilize DNA at 45 °C.
- Resuspend pellet in 10 μl labeling buffer and add to tube of dye without touching the dye.
- Repeat with another 10 μl labeling buffer.
- Vortex and spin down tube of dye and DNA.
- Cover tubes with aluminum foil and keep in dark at RT O/N to label.
- Repeat QIAquick Nucleotide Removal Kit Protocol. Perform with two differences:
– Add 200 μl PN buffer to probes and put labeled probes through columns 2x.
– Perform 3 washes with buffer PE in order to wash away any unattached dye before elution. - After elution obtain concentration via nanodrop. The labeling efficiency is typically ~0.25 pmol/ng of single-stranded DNA.
3. Coverslip Preparation
- Place coverslips on slides in Plasma-preen vacuum chamber (http://www.plasmapreen.com/) (the closer to the center the better).
- Put vacuum chamber in microwave and make sure it is sealed.
- Turn on pump first then turn on vacuum only once pump is started.
- Turn on microwave, and stop 5 sec after plasma is visible.
- Turn off vacuum then pump.
- Pull out vacuum chamber and remove coverslips with forceps (those that have fallen need to be cleaned again).
- Place coverslips cleaned side up in 12-well plates.
4. Fixation Procedure
- Grow yeast to an OD600 of around 0.1-0.2 in minimal media. 10 ml of cells yields enough for ~10 separate hybridizations.
- Add 1/10 volume 37% formaldehyde directly to the growth media (10 ml of culture + 1 ml 37% formaldehyde) and let sit for 45 min.
- Wash 2x with 1 ml ice cold Buffer B in a microcentrifuge tube (can spin at 13,000 rpm for 1 min).
- Add 1 ml of spheroplasting buffer.
- Incubate at 37 °C for 15 min. Check cells every few minutes under microscope until most cells are black (i.e. not phase-bright).
- Wash 2x with ice cold Buffer B, spinning at low speed (~3,500 rpm).
- Add 1 ml of 70% EtOH, resuspend gently and leave overnight at 4 °C (can store indefinitely at -20 °C).
5. Hybridization Procedure
- Prepare the hybridization solution: to 100 μl of hybridization buffer, add 1-3 μl of probe, then vortex and centrifuge. Singer probes use 8-10 ng total per probe set (i.e. per gene). We have imaged up to 3 genes simultaneously with 3 different labeled probe sets.
Be sure to warm the hybridization solution to room temperature before opening it.
For Stellaris probes, it is recommended to start 4 separate hybridization reactions by adding 1 μl each of 1:10, 1:20, 1:50 and 1:100 working dilutions of probes to see which one is optimal. Working dilutions of Stellaris probes are prepared in Hybridization buffer.
- Centrifuge (for all subsequent steps; 3,500 rpm; 5 min) the fixed cells (e.g. 200 μl) and aspirate away the ethanol.
- Gently resuspend in 1 ml wash buffer that contains the same percentage formamide as the hybridization buffer. Let stand for 2-5 min.
- Centrifuge sample and aspirate wash buffer, then add hybridization solution. Incubate in the dark with gentle shaking, O/N at 37 °C.
Note: the following procedure is for applying/imaging cells on coverslips. For washing/imaging cells in 96-well plates, including alternative reactive oxygen species scavenger solution see http://www.biosearchtech.com/stellarisprotocols.
- The next day, or in advance, clean (see procedure) and treat cover slips with 150 μl 0.01% Poly-L-Lysine for 5 min. Aspirate, let dry, wash 3x with dH2O and let dry.
- In the morning, add 1 ml of wash buffer to the sample, gently resuspend, centrifuge and aspirate, then resuspend in another 1 ml of wash buffer and incubate at 37 °C for 30 min.
- Wash with 2X SSC + 0.1% Triton X-100 at RT 15 min on shaker.
- Take mounting medium out of freezer to allow it to come up to RT before mounting.
- Wash with 1X SSC at RT 15 min on shaker.
- Dilute DAPI into PBS (0.1 μg/ml final) and resuspend cells in 150 μl.
- Place solution onto cleaned/poly-lys-treated cover slips in 12-well plate; at least 30 min undisturbed.
- Remove solution (you may place it on a spare cover slip as a backup) and wash 3x with 1 ml 1X PBS.
- Place 3 μl of Prolong Gold mounting medium onto a slide (Invitrogen P36934). Do this one at a time if you have multiple cover slips to avoid drying of mounting medium.
- Add ~0.5 ml ethanol to coverslip in 12-well plate, remove the cover slip and air dry while holding with tweezers.
- Place cover slip cell-side down onto mounting medium and let harden several hours or O/N in the dark.
- Seal the edges with nail polish and proceed to imaging.
6. Imaging of Cells with Olympus IX-81 Inverted Microscope Overview
- For image acquisition we utilize Slidebook (intelligent-imaging.com) software and a 100X, 1.45 NA, TIRFM oil objective. For serial GFP, DAPI, Cy3, Cy3.5 and Cy5 imaging: Chroma filter sets (see reagents).
- Use DAPI filter to find and focus cells.
- Set this as the reference point.
- Take a z-stack of images around the reference point where the total distance is 5 μM with 0.2 μM size (25 planes). Repeat for each dye channel (i.e. each probe set).
- Export as a 16-bit TIFF for image analysis.
7. Image Analysis Overview (Singer Probes)
Below we provide an outline of computational methods we use for analyzing FISH images in MATLAB. The relevant MATLAB functions used are bracketed on the right. The algorithms and thresholds are currently tuned for data from Singer style probes. Using Stellaris style probes will require some adjustment, particularly to the final filtering step (7.8).
Cell Identification15
- Separate cells from background using a global threshold on DAPI images16 [graythresh].
- Identify nuclei using extended maxima function [imextendedmax].
- Segment cells using nuclei as seeds for a watershed algorithm [watershed].
Find spots in each fluorescence channel
- Perform tophat transformation to normalize background and improve signal to noise ratio [bwmorph].
- Find maxima to identify the in-focus layer (in the z-plane) for each spot [imregionalmax].
- Filter potential spots using a linear fit model to a radial gradient.
Measure spot intensity and filter single vs. multiple probe signals
- Fit a 2D Gaussian profile to the spot in the previously identified in-focus layer and estimate intensity17.
- Filter out weak spots using a threshold (based on histogram). For Singer type probes this is important, but is less so for Stellaris probes.
- Count spots in each cell.
Representative Results
Figure 3 shows typical histograms computed from FISH images and used to determine the number of mRNAs present in single cells. An important advantage of microscopy-based RNA quantification is that one can obtain information on the localization of transcripts. For example, we used FISH to identify mRNAs in single cells with an inducible CBF1 allele (Figure 4). Because many mRNA molecules are present at the site of transcription, we are able to identify the presence and location of transcription sites within the nucleus.
By utilizing different dyes to label mRNAs of different genes, one can quantify multiple mRNA species in the same cells. To demonstrate this, yeast cells were incubated in the presence of α-factor and sorbitol. FUS1 transcription (Quasar670 dye, red) is induced by α-factor. STL1 transcription (Quasar 570 dye, green) is induced by increases in extracellular osmolarity (Figure 5). Figure 4 is an example of FISH with the Singer probes. Figure 5 is an example of FISH with the Stellaris probes.
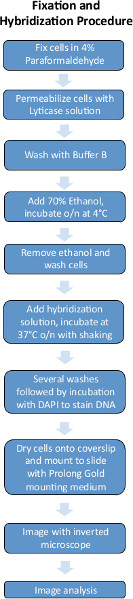
Figure 1. Schematic of FISH experimental procedure. Click here to view larger figure.

Figure 2. Schematic of image analysis pipeline. The final spots are determined in the rightmost figure.
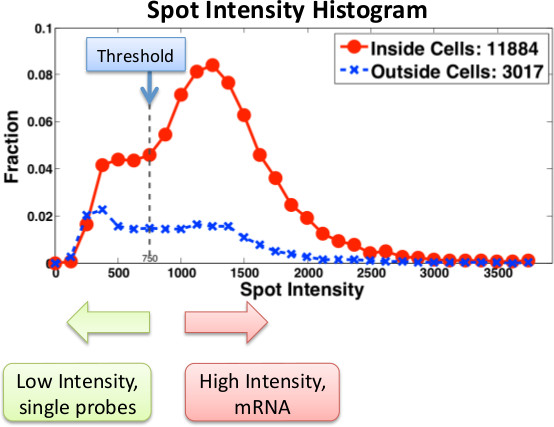
Figure 3. Histogram of spot intensities for a particular gene using Singer probes. Probe intensities are computed both inside (red) and outside (blue) cells. Low intensity spots are either noise or single probes. Real mRNA messages are labeled with multiple probes. When using Stellaris probes, single probes are less detectable and thus the thresholds and filtering must be adjusted accordingly.
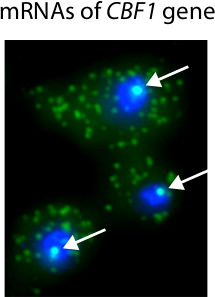
Figure 4. Representative results of Singer FISH procedure. In this experiment, CBF1 transcription is activated with an inducible promoter18. Nuclei are stained blue with DAPI. CBF1 mRNAs are tagged with Cy3-labeled probes. White arrows highlight the presence of CBF1 transcription sites in the nucleus. Single mRNA transcripts are visible in the cytoplasm.
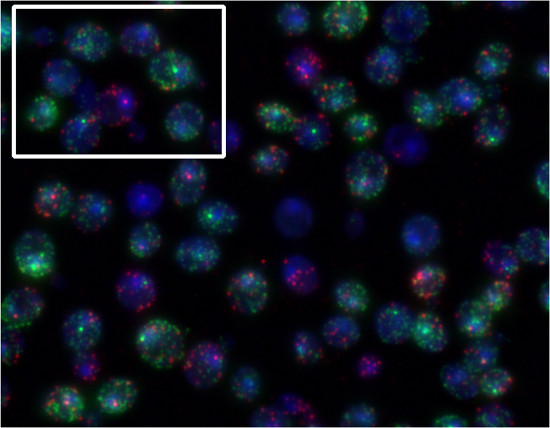
Figure 5. Representative results of Stellaris FISH procedure. MATa yeast cells were simultaneously exposed to 30 ng/ml α-factor and 0.75 M sorbitol for 10 min and were simultaneously probed for FUS1 (Quasar 670, red) and STL1 (Quasar 570, green) transcripts. In the highlighted box, we can see one cell responding only to the pheromone (FUS1 start site, red) and another predominantly responding to sorbitol (STL1 start site, green).
Discussion
To date, FISH has primarily been a low-throughput method. The use of Cy3, Cy3.5, and Cy5 dyes limits the number of genes one can investigate in single cells to three at a time. Some additional probes have been developed (Stellaris) but the number of distinguishable probes is still at most seven. To circumvent this limitation, combinatorial labeling strategies using multiple fluorophores have been used to create barcodes for different mRNA species19,20. Most recently, Lubeck and Cai used optical and spectral barcoding to quantify 32 different species simultaneously with FISH in single yeast cells19. One limitation of this recent combinatorial approach is it requires the use of super-resolution microscopy. The analysis needed to distinguish the barcoded probes is also quite complex.
We have found that Cy3 and Cy3.5 are preferable to Cy5 for FISH experiments. One of the limitations of the Cy5 dye is its sensitivity to photobleaching. However, Stellaris has recently developed Cy5 variants that are advertised as more resistant to photobleaching, and may alleviate this technical issue. It also worth noting that FISH is an expensive method to implement and that both Singer and Stellaris probes typically cost $700 – $1,000 per probe set, although prices for commercially available probes should decrease in the future. Sparing of reagents and efficient labeling brings Singer probes down to the lower range in price.
One of the major technical challenges is the separation of single versus multiple probe spots, which requires the implementation of sophisticated spot-determining algorithms. This can take extensive manual review to tune image analysis parameters for specific experimental setups. An outline of our computational pipeline with relevant MATLAB functions is provided in Section 7 of the protocol. This issue is somewhat alleviated by the Stellaris probes which have only one label per probe. It therefore requires the colocalization of multiple probes to see a signal.
Because FISH necessitates fixing cells, it does not facilitate tracking individual cells over time. Previously, we used FISH snapshot data to reconstruct the dynamics of gene expression in individual metabolically cycling yeast populations6. Metabolic cycling is observed in pre-starved, continuous cultures, and is characterized by population-wide collective oscillations in oxygen consumption. These oscillations are associated with genome-wide oscillations of transcripts that occur for half of all yeast genes at different phases of oxygen consumption. We sought to determine if metabolic cycling was present in unsynchronized continuous yeast cultures. If present, transcripts that are anti-correlated in synchronous populations should also be anti-correlated in unsynchronized single cells, and vice versa for correlated transcripts.
To reconstruct dynamics of mRNA production in time, the observed snapshot data must be compared to what is expected from a model of the underlying behavior. There are theoretical limitations to when such “snapshots” of gene expression data can be used to determine the underlying gene expression dynamics and which kinds of models can be distinguished21. For the metabolic cycle data, rather than directly showing the presence of temporal oscillations, statistical measurements were implemented to substantiate that there is indeed a cell autonomous oscillatory program consistent with bulk microarray measurements.
Divulgations
The authors have nothing to disclose.
Acknowledgements
This research was supported by grants GM046406 (to D.B.) and by the National Institute of General Medical Sciences Center for Quantitative Biology (GM071508). R.S.M. acknowledges funding from the NSF Graduate Research Fellowship. M.N.M. is supported by a Lewis-Sigler Fellowship. We would like to acknowledge members of the Botstein lab for helpful discussions and the former members Allegra Petti and Nikolai Slavov for their contributions to the metabolic cycle project. We thank Daniel Zenklusen and Robert Singer for getting us started with the FISH method.
Materials
| Name of the Reagent | Company | Catalogue Number | Comments |
| Vanadyl Ribonucleoside Complex | NEB | S1402S | |
| Lyticase | Sigma | L5263 | |
| E. coli tRNA | Roche | 1010954001 | |
| BSA (RNase free) | Ambion | ||
| Beta-mercaptoethanol | Fisher | 03446l | |
| DAPI, dilactate | Sigma | D9564 | |
| PBS 10X (RNase free) | Ambion | AM9624 | |
| Triton X-100 | Shelton Scientific | ||
| Dextran sulfate | Sigma | D6001 | Or equivalent |
| Saline-sodium citrate (SSC) 20X | VWR | 82021-484 | |
| Formamide (deionized) | Ambion | AM9342 | |
| Nuclease-free water | Ambion | AM9932 | |
| Alpha-D-glucose | Sigma | 158968 | For GLOX solution |
| 1 M Tris-HCl, pH 8.0 | Ambion | AM9855G | |
| 100% Ethanol | |||
| Glucose oxidase | Sigma | G0543 | For GLOX solution |
| Catalase | Sigma | C3155 | For GLOX solution |
| Concanavalin A | MP Biomedicals | 150710 | |
| Polylysine (0.01%) | Sigma | P8920 | |
| Coverslips | Warner Instruments | Cs-18R15 | |
| Prolong Gold Mounting Medium | Invitrogen | P36934 | |
| QIAquick Nucleotide Removal Kit | QIAGEN | 28304 | |
| FISH Probes | Biosearch Technologies | Custom order for your desired mRNA sequence | |
| Glass bottom 96-well plates | Nunc | 265300 | Alternative to coverslips |
| 12-well plates | BD Falcon | 351143 | |
| Cy3, Cy3.5, Cy5 dyes | GE Healthcare | monofunctional NHS-ester | |
| EQUIPMENT | |||
| Plasma-Preen I Cleaner | Terra Universal | 9505-00 | Controller (Cat #9505-17 optional) |
| Vacuum Pump | Alcatel | 205SDMLAM | For operating Plasma-Preen |
| Widefield Fluorescence Microscope | Olympus | IX81 | Or equivalent |
| 100X objective | Olympus | 1-UB617R | |
| Light Source | X-Cite | XCT 10-A | Or equivalent |
| Filter Sets | Chroma | U-NSP100V2-SPR, U-NSP101V2-SPR, U-NSP102V2-SPR, U-NSP103V2-SPR,U-NSP104V2-SPR. | |
| Cooled CCD or EMCCD Camera | Hamamatsu | C4742-98-24ER |
References
- Femino, A. M., Fay, F. S., Fogarty, K., Singer, R. H. Visualization of single RNA transcripts in situ. Science. 280, 585-590 (1998).
- Gall, J. G., Pardue, M. L. Formation and Detection of Rna-DNA Hybrid Molecules in Cytological Preparations. Proceedings of the National Academy of Sciences of the United States of America. 63, 378 (1969).
- Jones, K. W. Chromosomal and Nuclear Location of Mouse Satellite DNA in Individual Cells. Nature. 225, 912 (1970).
- Bauman, J. G., Wiegant, J., Borst, P., van Duijn, P. A new method for fluorescence microscopical localization of specific DNA sequences by in situ hybridization of fluorochromelabelled RNA. Exp Cell Res. 128, 485-490 (1980).
- Singer, R. H., Ward, D. C. Actin gene expression visualized in chicken muscle tissue culture by using in situ hybridization with a biotinated nucleotide analog. Proc. Natl. Acad. Sci. U.S.A. 79, 7331-7335 (1982).
- Silverman, S. J., et al. Metabolic cycling in single yeast cells from unsynchronized steady-state populations limited on glucose or phosphate. Proc. Natl. Acad. Sci. U.S.A. 107, 6946-6951 (2010).
- Trcek, T., Larson, D. R., Moldon, A., Query, C. C., Singer, R. H. Single-molecule mRNA decay measurements reveal promoter- regulated mRNA stability in yeast. Cell. 147, 1484-1497 (2011).
- Bertrand, E., et al. Localization of ASH1 mRNA particles in living yeast. Mol Cell. 2, 437-445 (1998).
- Gandhi, S. J., Zenklusen, D., Lionnet, T., Singer, R. H. Transcription of functionally related constitutive genes is not coordinated. Nat. Struct. Mol. Biol. 18, 27-34 (2011).
- Itzkovitz, S., et al. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nature Cell Biology. 14, 106-U193 (2012).
- Raj, A., Rifkin, S. A., Andersen, E., van Oudenaarden, A. Variability in gene expression underlies incomplete penetrance. Nature. 463, 913-U984 (2010).
- Raj, A., Tyagi, S. Detection of individual endogenous RNA transcripts in situ using multiple singly labeled probes. Methods Enzymol. 472 (10), 365-386 (2010).
- Trcek, T., et al. Single-mRNA counting using fluorescent in situ hybridization in budding yeast. Nature Protocols. 7, 408-419 (2012).
- Raj, A., vanden Bogaard, P., Rifkin, S. A., van Oudenaarden, A., Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nature Methods. 5, 877-879 (2008).
- Otsu, N. A Tlreshold Selection Method from Gray-Level Histograms. IEEE Transactions on Systems, Man and Cybernetics. 9, 62-66 (1979).
- Thompson, R. E., Larson, D. R., Webb, W. W. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 82, 2775-2783 (2002).
- McIsaac, R. S., et al. Fast-acting and nearly gratuitous induction of gene expression and protein depletion in Saccharomyces cerevisiae. Molecular biology of the cell. 22, 4447-4459 (2011).
- Lubeck, E., Cai, L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nature Methods. 9, 743-U159 (2012).
- Levsky, J. M., Shenoy, S. M., Pezo, R. C., Singer, R. H. Single-cell gene expression profiling. Science. 297, 836-840 (2002).
- Wyart, M., Botstein, D., Wingreen, N. S. Evaluating Gene Expression Dynamics Using Pairwise RNA FISH Data. Plos Computational Biology. 6, (2010).
