Modern gas-phase molecular physics and physical chemistry experiments often use supersonic expansions of target molecules to produce rotationally cold molecular samples within a molecular beam. However, even at low rotational temperatures of 1 K, which can routinely be achieved using supersonic expansions, large molecules can still remain in multiple conformations within the beam1. Similarly the production of molecular clusters in a beam source does not result in a single species, but rather in the formation of a "cluster soup", containing many different cluster stoichiometries, as well as remaining pure parent molecules. This makes the study of these systems with novel techniques such as imaging of molecular orbitals2, molecular-frame photoelectron angular distributions3-5 or electron6-10 and X-ray diffraction11-13 difficult, as these require pure, consistent, and homogenous samples in the gas-phase.
While several methodologies are now available to separate different conformers of charged species in the gas-phase (e.g. ion mobility drift tubes14,15) and charged clusters are easily separated by their mass-to-charge ratio, these techniques are not applicable to neutral species. We have recently demonstrated that these issues can be overcome with the use of an electrostatic deflection device16,17, allowing the separation of molecular conformers as well as clusters and the production of rotationally cold molecular beams.
The use of electrostatic deflection is a classic molecular beam technique, the origins of which go a long way back18,19. First ideas of utilizing electrostatic deflection for the separation of quantum states were introduced by Stern in 192620. While early experiments were conducted on small molecules at high temperatures, we demonstrate the application of this technique to large polar molecules and clusters at low temperatures16,21.
Polar molecules experience a force inside an inhomogenous electric field (E) due to the spatial differences in potential energy. This force is dependent on the effective dipole moment, μeff, of the molecule and can be evaluated as
is dependent on the effective dipole moment, μeff, of the molecule and can be evaluated as
 (1)
(1)
As different molecular conformers typically posses different dipole moments and differing numbers of solvent molecules within a cluster lead to different cluster masses and dipole moments, these species will experience a different acceleration in the presence of a strong inhomogeneous electric field. The resulting Stark effect force from an inhomogeneous electric field can therefore be used for the separation of conformers and quantum states22. This is indicated in Figure 1, showing the calculated Stark curves for the J = 0,1,2 rotational states of the cis and trans conformers of 3-fluorophenol, respectively. This leads to large differences in μeff, as shown in Figures 1c and 1d, and hence a different acceleration is experienced by the two conformers in inhomogeneous electric fields. Therefore, an electrostatic deflection device can be used as a mass-to-dipole moment ratio (m/μeff) separator, in analogy to a mass spectrometer acting as a mass-to-charge ratio (m/z) filter23.
Furthermore, these techniques allow the separation of rotational quantum states24,25. As the ground rotational states (blue curves in Figures 1a and 1b) exhibit the largest Stark shift, these will be deflected most and can be spatially separated from molecules in higher J states17. The coldest part of a molecular beam can therefore be selected, significantly aiding in many applications, such as alignment and orientation of target molecules17, 26-28.
In this contribution we show how an electrostatic deflection device can be used to spatially separate different species of large polar molecules and clusters. Example data is presented for the production of a pure beam of an individual conformer and of a solute-solvent cluster of well-defined size and ratio. Specifically we present data on 3-fluorophenol, where a pure beam containing only the trans conformer is produced, and on indole-water clusters, where the indole(H2O)1 cluster can be spatially separated from water, indole, indole(H2O)2 , etc.
The electrostatic deflection technique has been successfully applied to the separation of structural isomers16 and neutral clusters21, as well as the production of rotational quantum state selected molecular samples31. We demonstrate this with representative results for the separation of cis and trans conformers of 3-fluorophenol, and size selected indole(H2O)n clusters.
3-Fluorophenol conformers were separated in a molecular beam from the supersonic expansion of 50 bars of helium. The individual species were probed via their distinctive REMPI resonances around 272 nm32. Due to its significantly larger dipole moment (see Figure 1), the trans conformer experiences a larger deflection following passage through the deflector and is spatially separated from the cis conformer and the carrier gas of the beam.
To characterize the molecular beam formed during supersonic expansion, a temporal profile is collected with the electrostatic deflector turned off, as shown in Figure 3. For comparison, a temporal profile of a neon seeded beam is also shown. For helium carrier gas we observe a temporal width of approximately 12 μsec full-width at half maximum (FWHM), typical for an expansion from an Even-Lavie valve under these operating conditions.
The spatial distribution of the molecular beam is monitored by translation of the REMPI laser relative to the molecular beam direction, and spatial profiles are shown in Figure 4. This shows the spatial extent of the cis (red trace) and trans (blue trace) conformers at two different deflection fields, created by applying a potential difference of 14 kV or 28 kV across the deflector. For comparison field-free profiles are shown in both plots by the magenta (cis) and cyan (trans) curves. These yield a spatial width of the molecular beam of about 2 mm and show that, without the deflector, both species are mixed within the beam. In the presence of a deflection field the trans conformer undergoes a significantly larger deflection than the cis conformer and can effectively be separated from the other species present in the beam, such that at a position of y = 3 mm a pure trans sample is created and can be utilized for further experiments.
Cluster separation is demonstrated by supersonic expansion of indole in a "wet" carrier beam of helium containing trace amounts of water, leading to the formation of molecular clusters of the type indolem(H2O)n. According to the literature and to ab initio calculations, the indole(H2O)1 cluster has a significantly larger dipole moment (4.4 D) than pure indole (1.96 D), water (1.86 D) or the indole(H2O)2 cluster, and should, therefore, be deflected the most21,33. All indole containing species can be selectively probed via REMPI around 283 nm24,35, utilizing the lowest energy allowed electronic-excitation transition of indole. As this resonant excitation step involves different frequencies depending on the solvation of indole, the detection is fully species selective. Spatial profiles of the molecular beam are shown in Figure 5, these have been recorded with a potential difference of 26 kV between the rod and trough electrode and are fully species selective for indole (blue), indole(H2O)1 (red) and indole(H2O)2 (green). The lines indicate simulated values; details of numerical simulations methods can be found in the literature17,21. For comparison a field-free (deflector grounded) spatial profile is shown by the black curve. As expected the 1:1 cluster of indole and water experiences the largest deflection and at a position of y = 2-3 mm a pure beam of indole(H2O)1 is created. To highlight the effect of the deflector on the spatial molecular beam profile, the inset in Figure 5 shows the molecular beam density of indole(H2O)1 as a function of potential difference applied across the deflector. It indicates that as the field strength is increased, the coldest part of the molecular beam experiences an increasing deflection, while the warmer constituents experience a significantly smaller spatial separation and some density remains at the original position. This furthermore highlights the selection of the coldest part of the molecular beam.
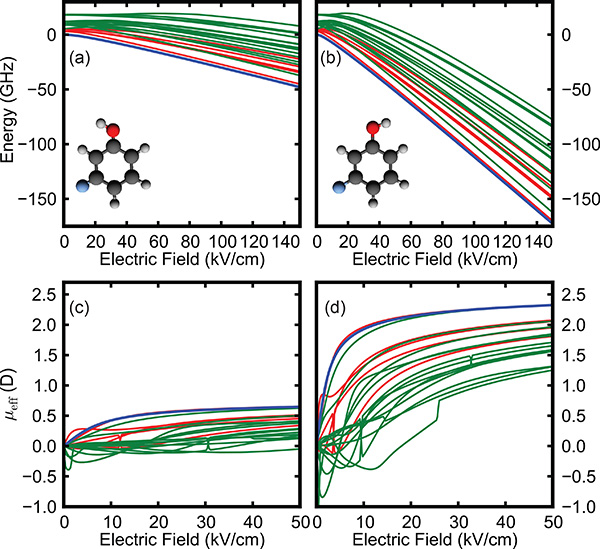
Figure 1. Calculated Stark energies E (top) and effective dipole moments μeff (bottom) for the cis and trans conformers of 3-fluorophenol. The blue line corresponds to the J = 0 rotational ground state, the red lines to J = 1 and green to J = 2 states. The deflection experienced is proportional to μeff/m (Equation 1). Hence, lower rotational quantum states, which exhibit larger μeff, experience a larger deflection and, therefore, can be separated. Equally, the significantly larger μeff for the trans conformer leads to a greater spatial deflection following passage through the electrostatic deflector.
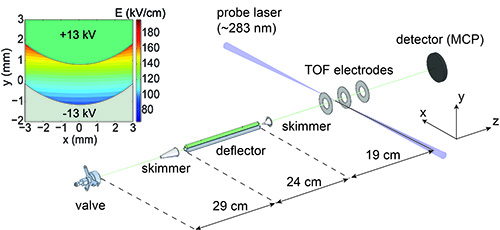
Figure 2. The experimental setup, consisting of a pulsed valve creating a supersonic expansion of target molecules, an electrostatic deflector and a detection region with time-of-flight mass spectrometer. The inset shows the inhomogeneous electric field created inside the deflector for voltages of ±13 kV applied to the rod and trough, respectively.
Click here to view larger image.
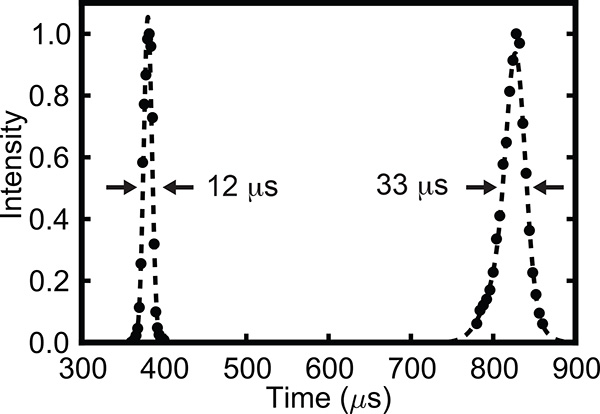
Figure 3. Temporal profile of the molecular beam for helium (at 380 μsec) and neon (at 826 μsec) carrier gas. The temporal width of the beam is approximately 3% and 4% of the total flight time for helium and neon, respectively.
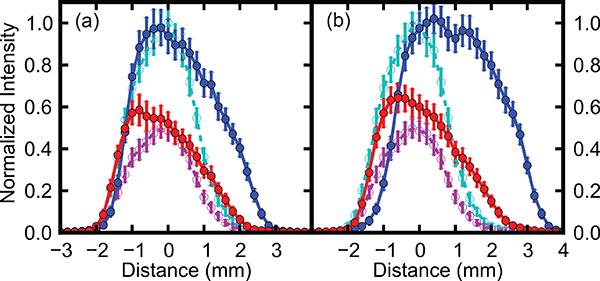
Figure 4. Spatial profiles of the molecular beam containing 3-fluorophenol, probed selectively for cis (red) and trans (blue) conformers, with the deflector at potential differences of (a) 14 kV and (b) 28 kV. For comparison the field free profile (deflector at 0 kV) is shown in both plots by magenta and cyan traces (cis and trans respectively).
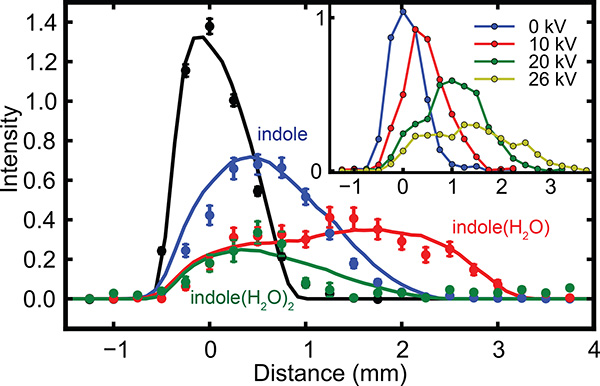
Figure 5. Spatial profiles of indole (blue), indole(H2O)1 (red) and indole(H2O)2 (green) for a deflector potential of 28 kV. Shown for comparison is the field-free profile of indole (black). Solid lines in the main panel indicate simulations. Shown inset are the measured spatial profiles for indole(H2O)1 at various potential differences applied across the deflector.
