Figure 1A shows a WF image of resting Cal-520 fluorescence in human neuroblastoma SH-SY5Y cells also loaded with ci-IP3. Exposure of these cells to a 100 msec UV flash to photo-release i-IP3 elicited transient Ca2+ puffs at discrete sites (noted by the white circles in Figure 1A). Fluorescence traces measured at these sites showed a rapid rising phase owing to transient openings of IP3Rs, followed by a much slower falling phase (Figure 1B and 1C) as Ca2+ slowly diffused away from the release site. Red highlighted events in Figure 1B are Ca2+ signals that were determined by the algorithm to have originated from the selected site. Non-highlighted events represent contaminating Ca2+ signals diffusing from closely adjacent sites.
To achieve higher-resolution imaging of local Ca2+ signals, cells may be further loaded with the slow Ca2+ buffer EGTA. This chelates Ca2+ ions diffusing between puff sites and thus prevents the generation of global Ca2+ waves that would obfuscate the study of local Ca2+ puffs generated by individual clusters of IP3Rs. Furthermore, EGTA accelerates the collapse of the local Ca2+ microdomain but, owing to its slow binding kinetics, minimally perturbs local free Ca2+ and its binding to the fast Ca2+ indicator within a cluster. The net effect of EGTA is to ‘sharpen’ fluorescence records of puffs in space and time, without appreciably diminishing their amplitude. A further improvement in resolution arises by using TIRF to limit excitation of the Cal-520 fluorophores to the narrow (<100 nm) evanescent wave created by TIRF. The sum of both of these approaches is a technique that effectively monitors instantaneous Ca2+ release flux (Ca2+ current) rather than a leaky integral of Ca2+ after it is released through the IP3Rs and slowly diffuses away from the release site and is ultimately sequestered 9. Figure 1D shows the TIRF ‘footprint’ of an SH-SY5Y cell when Cal-520 fluorophores were only excited within a narrow distance into the cell from the coverglass/aqueous solution interface. Rapid binding of Ca2+ to the fast indicator Cal-520 dye within the attoliter cytosolic volumes created by TIRF illumination, in conjunction with EGTA to ‘mop’ up diffusive Ca2+, yields fluorescence signals that are sharpened in both time and space (Figure 1E and 1F).
The analysis of image stacks captured in this manner is greatly facilitated through a custom written algorithm developed in our lab running on the open-source software platform Python11. Figure 2A shows the opening dialog window in which the user enters parameters to be used for identification and subsequent analysis of image stacks. Following this, the user is prompted to select a black level to be subtracted from the entire image stack after which the identification and analysis algorithm runs. Figure 2C-F show, respectively, the subcellular sites of local Ca2+ release; activity at selected sites; individual events on an expanded timescale; and spatial profiles of representative events (see Figure 2 legend for details). Upon user review of identified sites, analyzed data for all events (see Table 1) can be exported by selecting the ‘Save to Excel’ function.
Figure 3 presents data illustrating both the improvement in resolution achieved by TIRF imaging in EGTA-loaded cells as compared to WF imaging in unloaded cells, and the power of the algorithm in identifying and analyzing local Ca2+ signals. Figure 3A shows a representative local event imaged by WF in a cell loaded only with Cal-520 and ci-IP3. For comparison, Figure 3B shows an analogous event but now imaged by TIRF microscopy in a cell further loaded with EGTA. Superimposed traces in Figure 3C and 3D, illustrate corresponding temporal (C) and spatial (D) profiles of events recorded by WF fluorescence without EGTA (black) and by TIRF microscopy with EGTA loading (red). Figure 3E plots mean measurements of puff amplitudes, decay times, and spatial width under these two conditions, determined using the algorithm to analyze approximately 44 (WF) and 88 (TIRF) events.
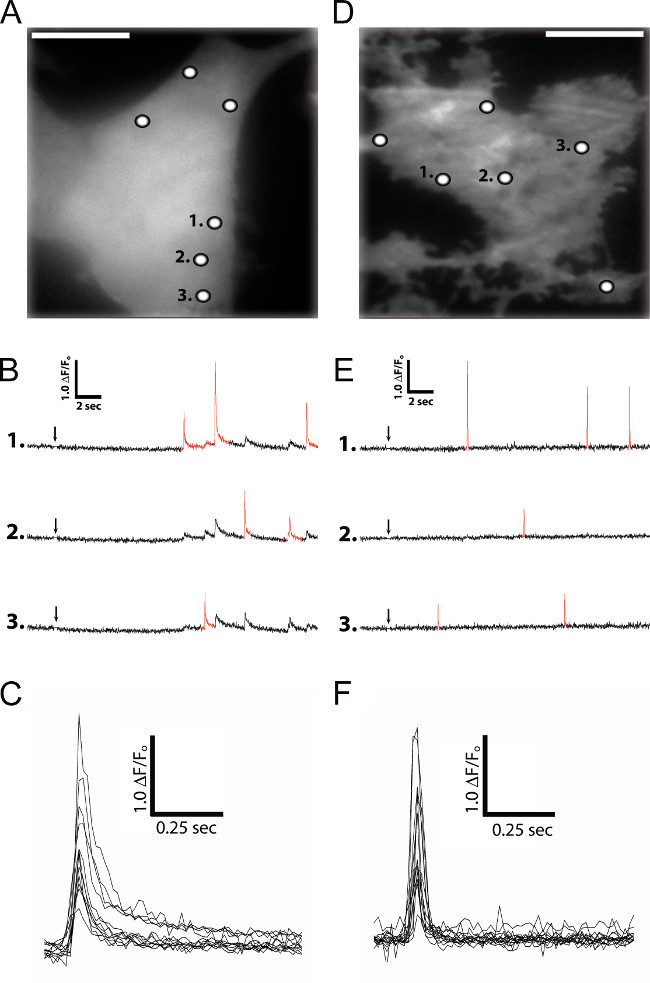
Figure 1: IP3-evoked local Ca2+ signals in SH-SY5Y cells loaded with Cal-520 and ci-IP3 imaged by wide-field (A through C) and TIRF (D through F) microscopy. (A) Monochrome image of resting Cal-520 fluorescence on which puff site locations are denoted by white circles; scale bar = 10 µm. (B) Traces illustrate puffs evoked at three different sites, numbered as in (A). Red highlighted events are puffs identified to have originated from that particular site. Arrow indicates time of UV flash. (C) Superimposed traces of representative Ca2+ puffs like those in (B), aligned and shown on an expanded scale. (D) Monochrome TIRF ‘footprint’ image of resting Cal-520 fluorescence in SH-SY5Y cells loaded with ci-IP3 and EGTA. White circles denote puff site origins. (E) Traces illustrate puffs evoked at three different sites numbered as in (D). (F) Superimposed traces of representative Ca2+ puffs like those in (E), aligned and shown on an expanded scale.
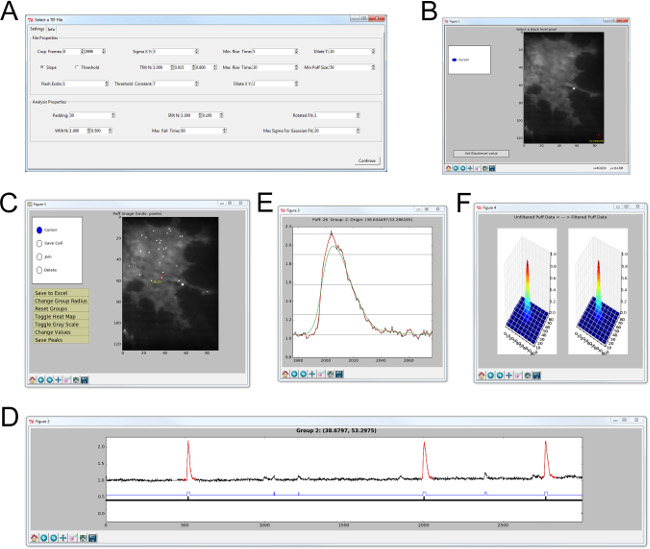
Figure 2: User interface of custom code designed to automatically identify and analyze local Ca2+ signals in SH-SY5Y cells imaged by TIRF microscopy. (A) Opening dialog box where identification and analysis parameters are entered. (B) The user selects a region of the image that contains no cells to define the black level that is subtracted from the all frames in the image stack. (C–F) Screenshots of the final/identification analysis windows. (C) Image of resting Cal-520 fluorescence on which are superimposed locations where Ca2+ transients were identified (white squares). The user can cycle between sites by moving the mouse over the image or pressing the cursor keys. (D) Window showing fluorescence ratio (ΔF/F0) trace at the selected event. Red-highlighted events are identified as having arisen at the site selected. The lower blue bar indicates events that exceed the threshold for detection at that site; events not highlighted red have been localized to an adjacent site. Clicking on the blue bar takes the user to that event site. Clicking on a red event with the mouse opens up two further windows showing the temporal evolution of the event on an expanded timescale (E) and spatial profiles (F) of the event averaged over its time course (left) together with the spatial profile of the fitted Gaussian (right). Please click here to view a larger version of this figure.
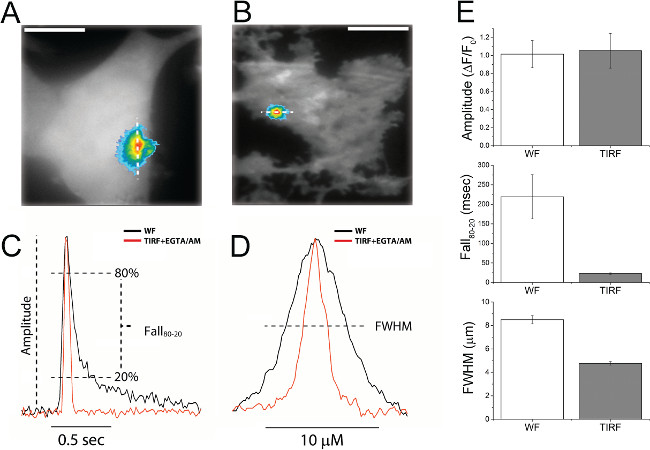
Figure 3: Improvement in temporal and spatial fidelity of puff imaging achieved by use of TIRF microscopy in conjunction with intracellular loading of EGTA. (A) snapshot image of a puff captured at time of peak amplitude, superimposed on a background image of resting fluorescence to show the cell outline. The image was obtained by WF microscopy of a cell not loaded with EGTA. The puff image is pseudocolored, with higher Ca2+ levels denoted by increasingly ‘warm’ colors. (B) Corresponding image of a puff imaged by TIRF microscopy in a cell loaded with EGTA. (C) Traces, WF (black) or TIRF + EGTA (red), show the corresponding change in Cal-520 fluorescence over time for the Ca2+ puffs shown in (A) and (B) above. Traces are normalized to the same peak amplitude and aligned to their peak time to facilitate comparison of their kinetics. Annotations illustrate measurements of Ca2+ event parameters (amplitude and fall time) quantified in (E). (D) Traces, WF (black) or TIRF+EGTA (red), show the fluorescence intensity of a line scan through the Ca2+ puffs shown in (A) and (B). Line scans are normalized to their peak amplitudes, and centered. The annotation shows measurement of full width at half maximal amplitude (FWHM), as quantified in (E). (E) Bar graphs showing mean peak amplitudes (top), fall times from 80% to 20% of peak amplitude (middle), and spatial width (FWHM) of puffs imaged by WF microscopy in cells without EGTA (open bars) and by TIRF microscopy in EGTA-loaded cells (grey bars). Data are presented as mean ± SEM with an n of at least 37 puffs for WF and 79 for TIRF. Please click here to view a larger version of this figure.
| A. | Group # | Event site identity number |
| B. | GroupX: | Mean sub-pixel x location of all events at a particular site |
| C. | GroupY: | Mean sub-pixel y location of all events at a particular site |
| D. | No. Events: | No. of events occurring at a site over the course of the experiment. |
| E. | Max Amp: | Maximum amplitude of an event at that particular site (ΔF/F0) |
| F. | X: | Sub-pixel x localization of event |
| G. | Y: | Sub-pixel y localization of event |
| H. | T_peak: | Time at which the event reaches peak amplitude (in frame number) |
| I. | Amplitude: | Amplitude of all events originating from each site (ΔF/F0) |
| J. | Sigmax: | X SD of Gaussian profile fitted to time course of event (in pixels) |
| K. | Sigmay: | Y SD of Gaussian profile fitted to time course of event (in pixels) |
| L. | Angle: | Angle of the long axis of the resulting elliptical function of Gaussian fitted to time course of event |
| M. | R20: | Time to rise to 20% of max amplitude (in frames) |
| N. | R50: | Time to rise to 50% of max amplitude (in frames) |
| O. | R80: | Time to rise to 80% of max amplitude (in frames) |
| P. | R100: | Time to rise to 100% of max amplitude (in frames) |
| Q. | F80: | Time to Fall to 80% of max amplitude (in frames) |
| R. | F50: | Time to Fall to 50% of max amplitude (in frames) |
| S. | F20: | Time to Fall to 20% of max amplitude (in frames) |
| T. | F0: | Time to return to pre-event baseline (in frames) |
Table 1: Output data from algorithm.
