As shown in Figure 1, synthesis of the Ir1 solvento-complex involved splitting of the chloride bridge in the parent dimer by precipitation of the chloride in the form of insoluble AgCl and association of water molecules to the metal center. The formation of Ir1 was confirmed by H1NMR and ESI. Additionally, UV-visible bands characteristic of metal ligand charge transfer and π-π* transitions were assigned to the spectrum, further validating the formation of Ir1. This solvated complex exhibits no emissive character when excited at 365 nm.
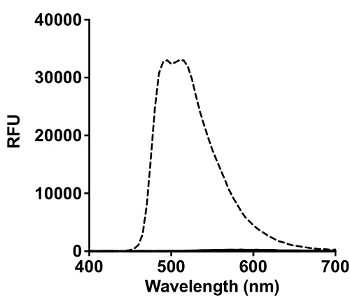
Figure 1: Synthesis and Characterization of Ir1. Chloride bridge splitting reaction of Dichlorotetrakis(2-(2-pyridinyl)phenyl)diiridium(III) to create the aquo complex Ir(ppy)2(H2O)2+ (Ir1). Upon addition of L-His to the aquo complex in aqueous buffer, luminescent signal is switched on.
Once the complex was synthesized and characterized, the amino acid selectivity was analyzed, as previously described using a similar iridium analog17. Figure 2A shows that only histidine elicited a signal response at 510 nm, when excited at 365 nm.
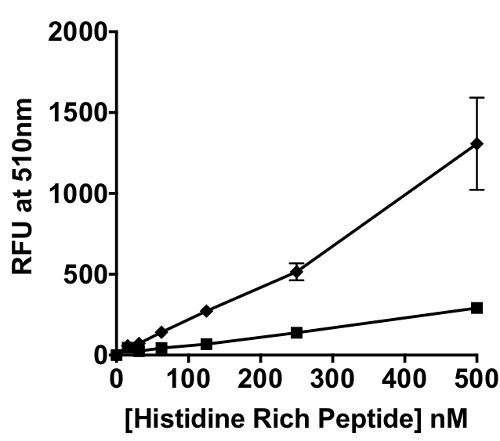
Figure 2: Amino Acid Selectivity and Titration of Ir1 with Histidine Rich Peptides. (A) Interaction of 200 µM of various amino acids (Cys, Ser, Asp, Glu, Phe, Lys, Arg, Tyr, Trp, His) with 50 µM Ir1 in HBS. Spectral scans were taken from 400-700 nm at an excitation wavelength of 365 nm in a black 96 well plate. Signal in relative fluorescence units (RFU) from all amino acids besides histidine (dashed black trace) was negligible. (B) Nanomolar concentrations of BNT-II (▪) and rcHRP-II (♦) were titrated with Ir1 in HBS. The histidine rich peptides were incubated with the probe for 15 min in solution before reading the emission at 510 nm after excitation with 365 nm light. Limit of detection was calculated as the value of x when y = 3σblank.
This “on-switch” of the phosphorescence of the iridium probe occurs because the singlet excited state (1MLCT/1LC) of the Ir(III) bioconjugate undergoes intersystem crossing to the triplet excited state (3MLCT), when histidine is coordinated to the metal center. This probe was applied to BNT-II a peptide mimic of the malarial biomarker Plasmodium falciparum Histidine Rich Protein II (pfHRP-II). In a titration of BNT-II with Ir1, a concentration dependent signal response was observed (Figure 2B). Recombinant HRP-II (rcHRP-II) also exhibited a similar response in solution. The limits of detection of BNT-II and rcHRP-II in solution were 54.8 nM and 12.8 nM, respectively. Using biolayer interferometry techniques, the KD of Ir1 binding to BNT-II immobilized on a Ni(II)NTA surface was found to be 2.05 µM (Figure 3).
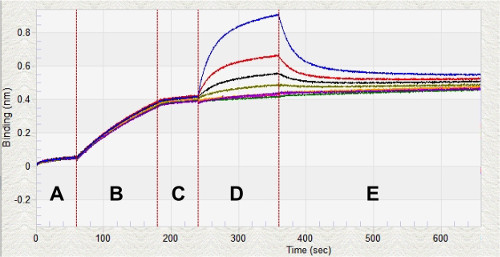
Figure 3: Real-time Kinetic Analysis of Ir1 with BNT-II. Biolayer interferometry for kinetic analysis of various concentrations of Ir1 binding to BNT-II on the surface of a Ni(II)NTA glass sensor. After equilibrating the sensors in KB (Region A), the sensors are loaded with 0.5 µM BNT-II (Region B). Once the peptide is loaded on the sensors, a baseline is established (Region C) prior to measuring the association of Ir1 to the BNT-II (Region D). After a period of association, the sensors are placed back into KB to measure dissociation (Region E). The whole kinetic analysis process takes less than 30 min.
When transitioning from a solution-based assay to a magnetic particle platform, the added complexity of the particle in the system had to be taken into account. It was known from previous work, that these particles efficiently bind the malaria biomarker pfHRP-II.24 Black fluorescent plates worked well for the solution assay describe above, but white plates were better suited for the on-particle detection platform, as seen in Figure 4A. In a white plate, the light is reflected back into the sample, thus allowing for better absorbance by the Ir1 bound to the surface of the particle. In the on-particle assay, the limit of detection of rcHRP-II was determined to be 14.5 nM (Figure 4B). The limit of detection for rcHRP-II in-solution and on-bead were statistically the same based on an unpaired t-test (p = 0.731).
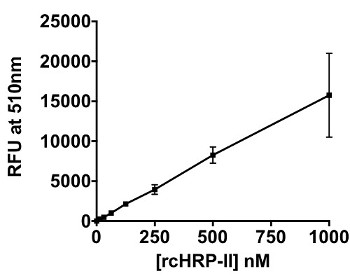
Figure 4: On-bead Detection of BNT-II and HRP-II. (A) Difference between the signal detected from Ir1 bound to BNT-II on the surface of 50 µm Ni(II)NTA magnetic agarose particles in a black 96-well plate (solid line) versus a white 96-well plate (dashed line). The particles were excited with 365 nm light, and emission was measured at 510 nm. (B) Titration of rcHRP-II immobilized on the magnetic particles and detected using Ir1. Limit of detection was calculated as the value of x when y = 3σblank.
Figure 5: Schematic Representation of the On-bead Detection of rcHRP-II with Ir1. General scheme of HRP-II binding to the surface of Ni(II)NTA particles and labeled with Ir1. The particles are incubated with a histidine rich peptide for 15 min. After this incubation period, the particles are washed with HBST using a magnet, in order to pull the particles out of solution. Finally, the peptide bound particles are incubated with Ir1 for 1 hr prior to reading the emission at 510 nm after excitation with 365 nm light.
