A schematic highlighting key steps in RNAseq Capture is shown in Figure 1. Four cancer cell lines with known mutations were used to demonstrate the effectiveness of the RNAseq Capture technique (K562 with ABL1 fusion, LC2 with RET fusion, EOL1 with PDGFRalpha fusion and RT-4 with FGFR3 fusion). The four samples were pooled together and sequenced with 2x 100 bp reads on a desktop sequencer, which generates FASTQ files. FASTQ files were run through an RNAseq analysis pipeline, which includes five main components: 1) quality control assessment, 2) alignment to human transcriptome, 3) gene expression quantification, 4) fusion calling, and 5) variant calling. The alignment file (BAM) is used to call single nucleotide variants and calculate gene expression. Fusions are called using fusion callers, such as TopHat Fusion (performing their own alignment) and the output is annotated using fusion detection software.
Comparison of gene expression from RNAseq and capture demonstrates enrichment of targeted transcripts by 10 to 1,000-fold using the capture method (Figure 2A). Additionally, Figure 2B shows an increase in the percent of reads mapping to the targeted transcript regions using capture compared to RNAseq. Assessment of quality control measures is represented in Figure 3. Capture and RNAseq perform equally in terms of alignment to the transcriptome (3A, 94% vs. 93%) and mean insert size (3B, 174 bp vs. 162 bp). Using the capture method, a higher percentage of exonic regions are sequenced (3C, 77% vs. 60%), and conversely a lower percentage of intronic regions are sequenced (3D, 4% vs. 20%). Total read counts per sample are depicted in 3E, and as expected, RNAseq generated over 50-fold more reads than capture. Finally, the percentage of rRNA sequences present in each sample was lower using the capture method when compared to RNAseq (3F, 4% vs. 15%).
Fusion detection output shown in Table 1 is generated with normalized fusion supporting reads. Capture RNAseq was successful in detecting fusions for all four cell lines. Comparison of single nucleotide variants called in overlapping regions of capture and RNAseq is displayed in Figure 4. This demonstrates a high concordance of variants between Capture and RNAseq within the target region.

Figure 1. Schematic of RNAseq Capture Steps. In this experimental demonstration, RNA is first depleted of ribosomal RNA, followed by chemical fragmentation and synthesis of complementary DNA (cDNA) using reverse transcriptase. Next, the cDNA is polyadenylated and ligated on both ends to platform-specific adaptors to generate a library. Only cDNA libraries with proper adaptors are then amplified by PCR. Libraries are then hybridized to custom oligonucleotide probes and captured using magnetic beads. This small amount of captured library must be amplified a second time to have enough for next generation sequencing. Multiple libraries can then be sequenced in parallel. Sequencing data is analyzed for RNA events of interest such as gene fusions, expression or mutations. Please click here to view a larger version of this figure.
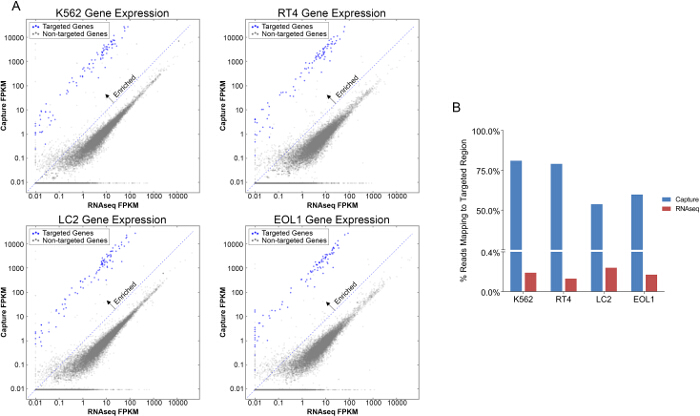
Figure 2. Comparison of Targeted Genes in Capture versus RNAseq. A, Gene expression comparison between Capture and RNAseq in four cancer cell lines K562, LC2, EOL1 and RT-4 measured by reads per kilobase per million mapped reads (FPKM)(Log scale). Targeted genes of interest are enriched (blue) compared to non-targeted genes (grey). B, Percentage of reads mapping to targeted region is increased in Capture versus RNAseq libraries in four cancer cell lines. Please click here to view a larger version of this figure.
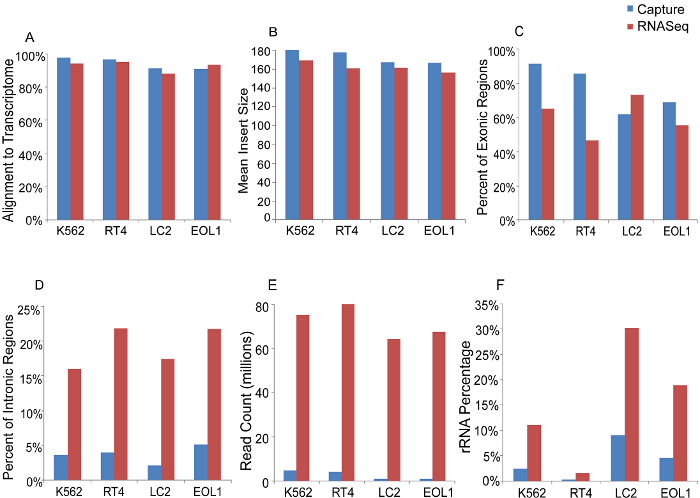
Figure 3. Sequencing Metrics of Capture versus RNAseq in Four Representative Cancer Cell Lines. A, Percentage of reads mapping to the transcriptome, B, Mean insert size of libraries. C, Percentage of reads in exonic regions. D, Percentage of reads in intronic regions. E, Total sequencing reads. F, Percentage of reads mapping to ribosomal RNA. Please click here to view a larger version of this figure.
| Cell Line | Fusion | Library Type | Total Reads | On Target Reads | Normalized Fusion Supporting Reads (NFSR) | ||
| TophatFusion | ChimeraScan | TRUP | |||||
| K562 | BCR-ABL | RNAseq | 150,300,482 | 279,438 | 0 | 438 | 0 |
| Capture | 9,341,148 | 7,566,087 | 598 | 343 | 0 | ||
| LC2 | CCDC6-RET | RNAseq | 128,861,790 | 307,566 | 0 | 97 | 0 |
| Capture | 12,320,692 | 10,314,284 | 71 | 44 | 6 | ||
| EOL1 | FIP1L1-PDGFRA | RNAseq | 135,321,406 | 225,222 | 0 | 0 | 170 |
| Capture | 9,317,418 | 7,680,818 | 143 | 0 | 7 | ||
| RT4 | FGFR3-TACC3 | RNAseq | 161,350,024 | 208,741 | 0 | 131 | 469 |
| Capture | 8,305,950 | 6,563,574 | 358 | 88 | 34 | ||
Table 1. Fusion Detection for Capture versus RNAseq of K562, LC2, EOL1 and RT-4. This Table displays four cancer cell lines and three different fusion detection algorithms, TopHat2, ChimeraScan, and TRUP utilized in this demonstration. This table demonstrates the ability to detect fusions with Capture using less than 10 million total reads compared to greater than 60 million reads utilized for RNAseq. Fusion supporting reads were calculated by dividing fusion supporting reads by kinase reads, multiplied by one million.
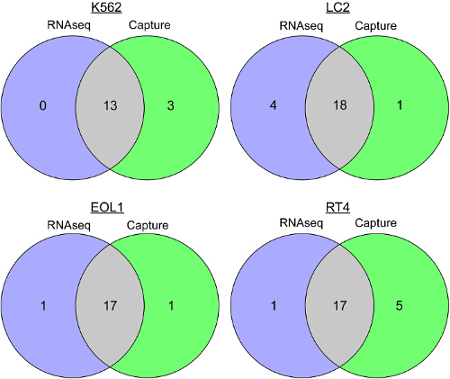
Figure 4. SNV calling for Capture versus RNAseq. These Venn diagrams show the number of Single Nucleotide Variants (SNVs) that were detected by Capture and RNAseq for each of four cell lines (K562, LC2, EOL1 and RT-4). This illustrates high concordance of SNVs between Capture and RNAseq within targeted-region: K562 (81.3%), LC2 (78.3%), EOL1 (89.5%) and RT-4 (73.9%). Please click here to view a larger version of this figure.
