This protocol represents an optimized method for recovering DNA and RNA from tissue cores, using modifications of a commercial extraction system designed for tissue sections. Optimization included the introduction of tissue homogenization, utilization of more potent Proteinase K for DNA extraction, and extension of tissue digestion time. Graphs and statistical analyses included 2-way ANOVA, linear regression and correlation.
Optimizations of Proteinase Digestion
The commercial kit included a room temperature stable proteinase K solution which was substituted with a more potent proteinase K, resulting in higher DNA yield (Figure 2A). To further increase the DNA yields, digestion was extended from 2 to 24 hr. No significant differences were seen between the two time points, but the 24 hr digestion appeared to provide more consistent yields across samples. However, further incubation to 48 hr did not further improve DNA recovery (Figure 2B; p = 0.74).
Typical DNA Recovery from FFPE Prostate Cancer Tissue Samples
Using the optimized protocol, RNA and DNA were co-extracted from 333 prostate cancer FFPET samples ranging from 3 to 14 years in sample age. From each sample, 3 tissue cores (average total tissue volume of 0.95 ± 0.13 mm3) were used as input. While there are other microfluidic based gel-electrophoresis methods which can estimate concentrations and provide evaluations of the size distribution of nucleic acids molecules, such methods do not provide reproducible nucleic acids quantification, and cannot distinguish between RNA and DNA as flourometrically-based assays do.22 And, because microfluidic based gel-electrophoresis results are not reliable for fragmented nucleic acids derived from FFPET,23 nucleic acid yields were measured fluorometrically (see reagent list for details). The average yield was 2,270 ng of RNA and 820 ng of DNA (Figure 3A). Approximately 90% of all FFPET samples analysed in this study yielded ≥100 ng of DNA and ≥ 500 ng of RNA. Interestingly, there was no significant correlation between the age of the FFPET sample and nucleic acid recovery (Figure 3B). Overall, RNA and DNA yields were correlated across samples (R2 = 0.39; p <0.0001), although more than twice as much RNA than DNA was recovered from each sample (Figure 3C).
As the pilot and optimization work was performed on prostate tissues, the next step was to investigate the performance of this protocol on a few additional types of archival tissue. Starting with surgically removed and autopsy FFPET samples representing benign liver (1 sample from 1 case), cancers of the brain (8 samples from 1 case), urinary bladder (2 samples from 2 cases), and breast (3 samples from 3 cases), the protocol yielded >100 ng of DNA and RNA from 90% of samples (Figure 3D). While nucleic acid yields were lower in autopsy tissues than in surgical tissues, representative results indicate that the protocol produces similar yields across cancers derived from different sites.
Assessment of RNA and DNA Integrity and their Representative Performance in Downstream Analysis
RNA expression analysis of 47 genes in 8 selected FFPET prostate cancer samples and a fresh PC-3 prostate cancer cell line sample (as a positive control) was performed using a commercial multianalyte gene expression platform that is optimized for FFPET. The mRNA counts in PC3 were typically higher than those from FFPET samples (Figure 4A). However, comparing relative expression of all genes, FFPET prostate cancer samples showed similar expression profiles to PC-3 RNA, indicating that both sources of RNA are suitable for RNA expression profiling.
To demonstrate performance of genomic DNA extracted with this protocol, bisulfite-converted DNA extracts from FFPET samples were amplified by methylation specific PCR (MSP).24 MSP analysis of ALU repetitive elements, highly methylated regions present in millions of copies in the human genome,25 was used as a genomic methylation control, and expected to show minimal variations between samples. As shown in Figure 4B, there was little to no variation seen between different samples in ALU MSP methylation levels. Further, MSP assays based on GSTP1, a gene known to be hypermethylated in prostate cancer but not in benign samples,26 showed no detectable amplifications in DNA from benign samples. As expected, lower qPCR cycle threshold values were detected in DNA from cancer tissues, indicating enrichment of methylated GSTP1 copies. The utility of nucleic acids recovered by this protocol was further tested in typical downstream assays, using nucleic acids recovered from benign liver and from a brain (post-mortem) and from two surgically removed breast cancer samples. Both RT-qPCR based expression and MSP assays performed well on breast cancer and liver FFPET, but the RT-PCR assay failed to amplify a highly expressed mRNA from the post-mortem brain tumor sample (Figure 4C), suggesting that RNA had degraded, likely due to delayed tissue fixation.
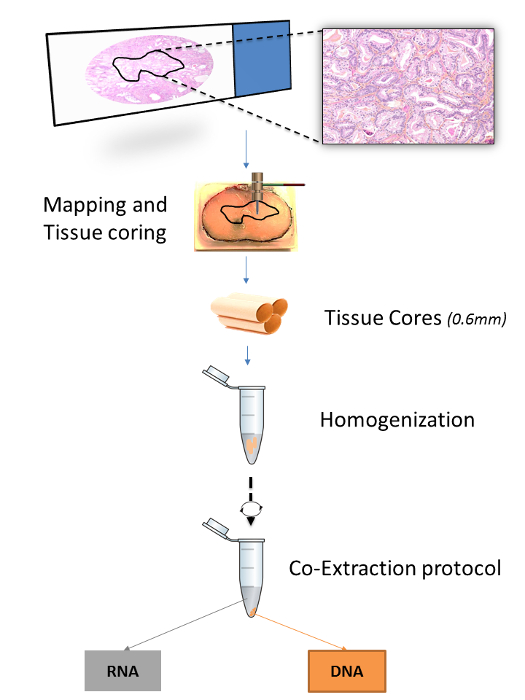
Figure 1: Overview of the Extraction Procedure for FFPET Samples. The figure illustrates how an area of interest in a tissue block is mapped based on histopathologic selection from a microscope slide. Three 0.6 mm tissue cores are then obtained from each tissue area by using biopsy punches, homogenized together and then subjected to extraction of both RNA and DNA. Please click here to view a larger version of this figure.
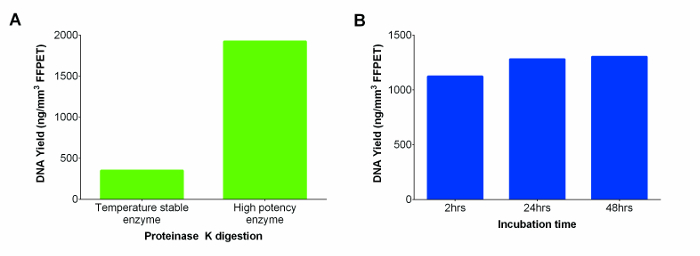
Figure 2: Nucleic Acids (DNA and RNA) yields in ng/mm3 of FFPET from two Proteinase K (Temperature Stable and High Potency Enzymes), and Tested across a Range of Incubation Times for the Latter. (A) Performance of Proteinase K from different suppliers. DNA extractions were performed on a representative FFPET sample using temperature stable enzyme supplied with the kit versus a more potent enzyme from another manufacturer. (B) Determining optimum Proteinase K incubation time to maximize the DNA yield. Performance of high concentration Proteinase K digestion was evaluated at three different incubation periods using 3 FFPET samples. Error bars represent standard error of mean (SEM). Please click here to view a larger version of this figure.
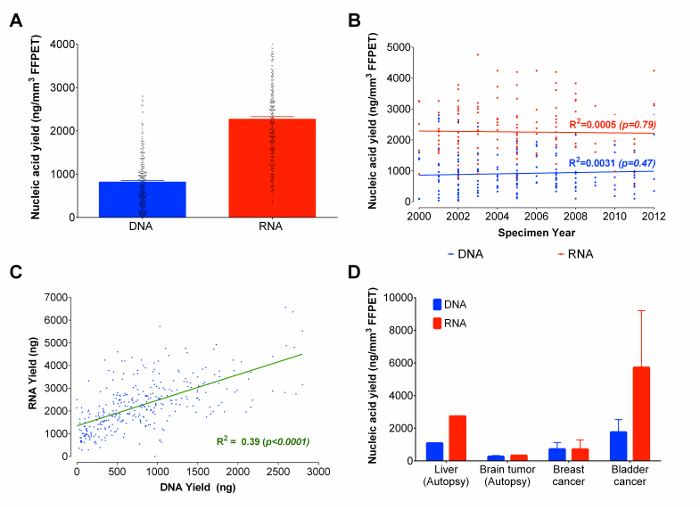
Figure 3: Nucleic Acids (DNA and RNA) yields in ng/mm3 of FFPET in Total, across Sample Years and Representative Tissue Types. (A) Total recovered nucleic acids from formalin-fixed paraffin-embedded tissue. The nucleic acids quantities presented are based on the extractions of 333 FFPET samples using the optimized protocol. (B) Correlation plot between recovered total DNA and RNA and the age of FFPET samples. The extracted FFPET samples used were obtained from the years 2000 to 2012. (C) Correlation between yields from concurrently extracted DNA and RNA from 333 prostate samples. There is a positive correlation between DNA and RNA yields. (D) Demonstration of the protocol using additional archival tissue types. The optimized protocol was used to extract nucleic acids from 14 cancer (breast, bladder and brain) and normal (liver) samples. Error bars represent SEM. Please click here to view a larger version of this figure.
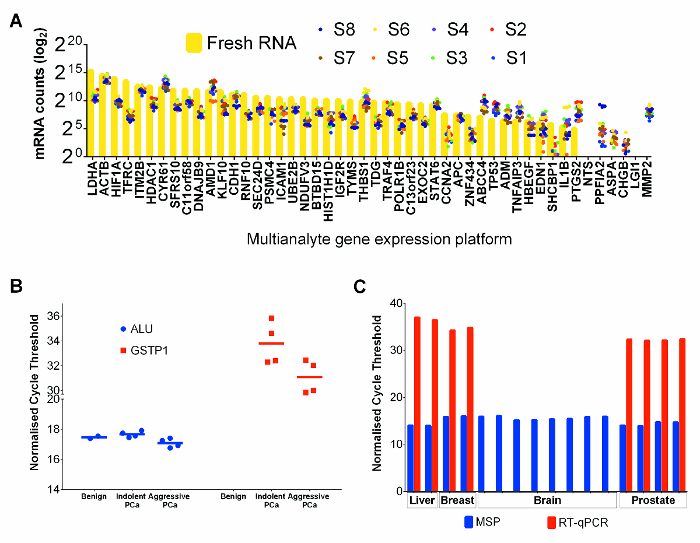
Figure 4: Performance of RNA and DNA Co-extracted from Tissue Cores in Downstream Applications. (A) mRNA counts for FFPE prostate cancer tissues and for fresh PC-3 cell line control. Each point represents the average of 3 technical replicates extracted separately. Fresh PC-3 cell line RNA values are illustrated by yellow bars and FFPET tissue values are represented by coloured dots. (B) Methylation specific PCR assays on DNA of FFPET prostate cancer samples. Cycle threshold values were obtained for 10 samples performed as expected using 50 ng/reaction of bisulfite converted DNA. ALU MSP assays from all FFPET samples had similar cycle threshold values (p >0.67). GSTP1 MSP assays showed higher methylation (lower cycle threshold) levels in prostate cancer than in benign prostate. (C) Assessment of DNA and RNA quality from additional tissue types. HPRT1 gene expression and Alu gene methylation assays were performed on nucleic acids (RNA and DNA respectively) extracted from normal liver (autopsy), and brain (autopsy) and breast cancers (all FFPET). Results from prostate are shown for comparison. Note: similar results were observed from each tissue type, except for failed mRNA amplification from one autopsy sample. Each point or bar represents a sample, and error bars represent SEM. Please click here to view a larger version of this figure.
| Tissue Cores | |||||||||||
| Tissue Input (mm3) | Extracted Nucleic Acid | Validation | Quality check: DNA | Quality check: RNA | |||||||
| (# of samples) | Age of Sample (years) | Total Yield (ng) | Fragment size (bp) | PCR | Total Yield (ng) | Fragment size (bp) | PCR | NanoString | |||
| Pikor et al. | 43 – 129 | DNA | No data | No data | No data | No data | |||||
| Montaser-Kouhsari et al. | 18 – 29.5 | RNA | 763 | 0-25 | 843 | No data | |||||
| This paper | 1.71 | DNA and RNA | >350 | 3-12 | 820 | 100 – 500 | 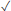 |
2270 | 100-500 | |
|
| Tissue Sections | |||||||||||
| Tissue Input | Extracted Nucleic Acid | Validation | Quality check: DNA | Quality check: RNA | |||||||
| (# of samples) | Age of Sample (years) | Total Yield (ng) | Fragment size (bp) | PCR | Total Yield (ng) | Fragment size (bp) | PCR | NanoString | |||
| Heikal et al. | 5 x 5 µm | DNA | 12 | 7-22 | 88-300 | 103-351 | |
||||
| Chung et al. | 1 x 20 um | RNA | 9 | >5 | 16,000- 23,000 | 100-200 | |
||||
| Antica et al. | 2 x 4 µm | RNA | 18 | No data | Unknown (621 ng/µl) | 80-202 + | |
||||
| Ghatak et al. | 5 x 5 µm | DNA and RNA | 5 | 1 | 14 256 | <1030 | |
16 000 | 109-400 + | |
|
| Hennig et al. | 1 x 10 µm | DNA and RNA | 210 | 1-25 | No data | No data | |
No data | No data | |
|
| Laser Capture Microdissection | |||||||||||
| Tissue Input (mm2) | Extracted Nucleic Acid | Validation | Quality check: DNA | Quality check: RNA | |||||||
| (# of samples) | Age of Sample (years) | Total Yield (ng) | Fragment size (bp) | PCR | Total Yield (ng) | Fragment size (bp) | PCR | NanoString | |||
| Snow et al. | 1-2 | DNA | 110 | 0-2 | 430 | No data | |
||||
Table 1: A comparison of published DNA and RNA extraction protocols for tissue cores, sections and laser capture microdissections. Also included are several molecular endpoints assessment of these methods using PCR and Nanostring assays.
