Assays for Studying the Role of Vitronectin in Bacterial Adhesion and Serum Resistance
Summary
This report describes protocols for characterizing interactions between bacterial outer membrane proteins and the human complement regulator vitronectin. The protocols can be used to study the binding reactions and biological function of vitronectin in any bacterial species.
Abstract
Bacteria utilize complement regulators as a means of evading the host immune response. Here, we describe protocols for evaluating the role vitronectin acquisition at the bacterial cell surface plays in resistance to the host immune system. Flow cytometry experiments identified human plasma vitronectin as a ligand for the bacterial receptor outer membrane protein H of Haemophilus influenzae type f. An enzyme-linked immunosorbent assay was employed to characterize the protein-protein interactions between purified recombinant protein H and vitronectin, and binding affinity was assessed using bio-layer interferometry. The biological importance of the binding of vitronectin to protein H at the bacterial cell surface in evasion of the host immune response was confirmed using a serum resistance assay with normal and vitronectin-depleted human serum. The importance of vitronectin in bacterial adherence was analyzed using glass slides with and without vitronectin coating, followed by Gram staining. Finally, bacterial adhesion to human alveolar epithelial cell monolayers was investigated. The protocols described here can be easily adapted to the study of any bacterial species of interest.
Introduction
Vitronectin (Vn) is an important human glycoprotein involved in maintaining homeostasis via regulation of the fibrinolytic system. Vn also functions as a complement regulator by inhibiting the terminal complement pathway during C5b6-7 complex formation and C9 polymerization. Several bacterial pathogens have been shown to recruit Vn to the cell surface as a means of resisting complement deposition1,2,3. In addition, Vn functions as a "sandwich" molecule between bacteria and host epithelial cell receptors, thereby promoting adherence and internalization of pathogens2,4,5. Binding of Vn to the bacterial cell surface is mediated by other currently unidentified proteins. Fully elucidating the functional role of Vn-binding in evasion of the hose immune response will therefore require identification of Vn-recruiting proteins.
The initial step in identifying Vn-binding proteins is to test whether a pathogen of interest can bind purified Vn. Flow cytometry is a convenient and straightforward method to determine whether Vn is bound to pathogen cells. In this study, we assessed the binding of Vn to various Haemophilus influenzae type f (Hif) clinical isolates6. The method described herein is quantitative and can be used to distinguish the binding capacity of a wide variety of bacterial strains. In a previous study, we characterized protein H (PH) of Hif as a Vn-binding protein7. Therefore, in the present study, the Vn-binding potential of wild-type (WT) Hif and Hif M10Δlph mutants were compared using the described protocols.
Once it is determined that a pathogen binds Vn, the second step is to characterize the surface proteome in order to identify potential Vn-binding proteins. A variety of approaches can be used for this purpose8,9, but these methodologies are not described in this report. The method most suitable for examining protein-protein interactions is to recombinantly express selected bacterial surface proteins in E. coli and purify by affinity chromatography. Here, we use PH and its molecular interaction with Vn to illustrate the method. Interactions between recombinant PH and Vn were characterized using an enzyme-linked immunosorbent assay (ELISA)7 and a recently developed label-free technique known as bio-layer interferometry (BLI)10,11. Whereas ELISAs can be used to confirm protein-protein interactions, BLI provides detailed data regarding the kinetic parameters of the interactions.
To study the functional role of Vn in bacterial adherence, two different assays can be utilized. The first assay described here is direct measurement of bacterial adherence to Vn-coated glass surfaces, whereas the second assay examines adherence to the surface of epithelial cells. For the first assay, glass slides were coated with Vn, and the binding of WT or mutant Hif strains was assessed by Gram-stain and microscopy. This technique readily distinguishes bacteria based on the ability to bind Vn12. Bacterial adhesion to mammalian cells was then analyzed by adding cultured bacteria onto a monolayer of type II alveolar epithelial cells; bacterial attachment was assessed by counting the number of colony-forming units (CFUs). Adhered and internalized bacteria can be distinguished in the presence or absence of Vn4,13.
The role of Vn acquisition in bacterial serum resistance was evaluated using a serum killing assay (i.e., serum bactericidal activity). To assess the significance of Vn acquisition in serum resistance, the bactericidal activity of Vn-depleted serum (VDS) was compared with that of normal human serum (NHS). The method used readily distinguishes Vn-binding versus non-binding bacteria based on serum resistance. We used this method to study the role of Vn in the serum resistance of several bacterial pathogens4,12.
Numerous methods have been reported for studying host-pathogen interactions. Here, we describe a set of protocols that can be easily adapted to the study of any pathogen in order to assess the role of Vn in pathogenesis. We tested these protocols using various pathogens, and Hif was chosen as an example for this report.
Protocol
1. Analysis of Vn as a Bacterial Surface Protein Ligand
- Detection of Vn-binding at the bacterial surface using flow cytometry
NOTE: In flow cytometry, we used side scatter and forward scatter to gate positive events. To examine the interactions with Vn, Hif clinical isolates (n=10)7 were selected together with E. coli BL21 (DE3) as a negative control (Figure 1A).- Culture Hif clinical isolates in brain-heart infusion (BHI) medium supplemented with 10 µg/mL NAD and hemin at 37 °C with shaking at 200 rpm. Use Luria-Bertani medium to culture E. coli14. Harvest Hif at mid-log phase (OD600 = 0.3)15, and resuspend the bacteria in phosphate-buffered saline (PBS), pH 7.2, containing 1% (w/v) bovine serum albumin (BSA) (blocking buffer; PBS-BSA). Adjust the suspension to 109 CFU/mL.
- Transfer the aliquots containing 5 x 106 CFU to 5 mL polystyrene round-bottom tubes (12 x 75 mm2) and add 1 mL of blocking buffer. Centrifuge the suspension at 3,500 × g at room temperature (RT)16 for 5 min to pellet the bacteria, then carefully aspirate to remove the supernatant without disturbing the pellet.
- Resuspend the bacterial pellets with 50 μL of blocking buffer containing 250 nM Vn and incubate the samples for 1 h at RT without shaking. After incubation, pellet the bacteria by centrifugation at 3,500 x g for 5 min, then wash the pellets three times using PBS and similar centrifugation steps.
- To the bacterial pellet, add 50 μL of primary sheep anti-human Vn polyclonal antibodies (pAbs) at 1:100 dilution in PBS-BSA. Incubate the suspension for 1 h at RT, then wash the bacteria three times with PBS to remove unbound antibodies (as described in step 1.1.3).
- Next, add 50 μL of PBS-BSA containing fluorescein isothiocyanate (FITC)-conjugated donkey anti-sheep pAbs (1:100 dilution) and incubate at RT for 1 h in the dark.
- Prepare a blank control by incubating bacteria with blocking buffer only and pAbs without Vn.
- Wash bacteria three times with 1 mL of blocking buffer and pellet the suspension by centrifugation, as described in section 1.1.2. Finally, resuspend the bacterial pellet with 300 µL of PBS and analyze by flow cytometry7.
- ELISA analysis of the interaction between recombinant PH and Vn
NOTE: Controls need to be included to exclude nonspecific binding. Human Factor H (FH) or C4b-binding protein (C4BP) are used as positive and negative controls, respectively.- Dilute each of the human proteins (Vn, FH, and C4BP) separately to 50 nM in Tris-HCl, pH 9.0 (coating buffer). Dispense 100 µL of protein solution into each well of a Polysorp microtiter plate. Close the plates with the lid and store at 4 °C overnight (16 h) to facilitate the immobilization of protein onto microtiter plate wells.
- Discard the solution from the microtiter plate by tilting upside down over the sink and wash the wells three times with 300 µL of PBS/well. Block the coated wells for 1 h at RT with PBS containing 2.5% (w/v) BSA (PBS-BSA).
- After removing the blocking solution, wash the wells three times with 300 µL of PBS containing 0.05% (v/v) Tween 20 (PBST) per well. Add 100 µL of 50 nM recombinant His-tagged PH to each sample well and incubate for 1 h at RT. In control wells, add only 100 µL of PBS-BSA.
NOTE: The lph gene encoding PH from Hif was amplified by PCR and cloned into the pET26b expression vector that adds a 6× His-tag at the C-terminus of the expressed protein. The recombinant vector was transformed into E. coli BL21(DE3) for expression. Ni-NTA resin was used to purify the recombinant protein15. - Discard the protein solution and remove the unbound proteins by washing the wells three times with 300 µL of PBST per well. Add 100 µL of PBS-BSA containing horseradish peroxidase (HRP)-conjugated anti-His pAbs (1:10,000 dilution) and incubate for 1 h at RT.
- Prepare 20 mM solution A by dissolving tetramethylbenzidine in a solution of 5% acetone and 45% methanol. To prepare solution B, dissolve 19.2 g of citric acid in 1,000 mL of H2O, adjust the pH to 4.25 by adding KOH, then add 230 µL of 30% H2O2. Store both solutions in the dark at RT. Just before use, mix 500 µL of solution B with 9.5 mL of solution A to prepare the ELISA detection reagent.
- Wash the wells three times with 300 µL of PBST per well and detect antigen-antibody complexes by adding 100 µL of ELISA detection reagent to each well.
- Add 50 µL of 1 M H2SO4/well to stop the reaction. Measure the optical density of the wells at 450 nm using a microplate reader.
- Study of the interaction kinetics of recombinant PH and Vn using BLI
- Immobilize human Vn on amine-reactive sensors using the amine coupling method, according to the manufacturer's guidelines11.
- Using PBS, serially dilute the ligand (recombinant PH) from 0 to 4 µM and transfer the resulting solutions to a 96-well black, flat-bottom microtiter plate. Run the experiment at 30 °C using a BLI instrument17.
- Load the data folder in the BLI data analysis software. Select the 'sensor selection' option. Then, select 'reference well' (Vn-coated sensor in PBS) for subtraction.
- Select 'align Y-axis to baseline' and select 'interstep correction and align to association'. Press 'process data' that will automatically open the analysis tab.
- In the analysis tab, select the 'association and dissociation' option under curve fitting. Select the model '1:1 binding and global fitting'. Press 'fit curve' and export fitting data17.
2. Characterization of The Role of Vn in Bacterial Adherence
- Study of bacterial adherence to Vn-coated glass surfaces
- Prepare a 2 µg/mL solution of Vn in PBS and pipet 10 µL of this solution onto a glass microscope slide as a single drop. Allow the drop to dry on the slide for 30 min at RT. Coat a slide with human serum albumin (HSA) as a negative control.
- Wash the protein-coated glass slides three times by dipping the slides for two seconds in a beaker containing PBS to remove excess uncoated protein. Add 20 mL of fresh Hif culture (described in step 1.1.1) into a sterile plastic petri dish and submerge the Vn-/HSA-coated glass slides in the culture medium. Incubate the dishes at 37 °C for 1 h with shaking at 20 rpm.
NOTE: Hif M10 and Hif M10Δlph were grown in liquid BHI medium or on chocolate agar plates. The medium for the lph mutant was supplemented with 10 µg/mL kanamycin15. - After incubation, remove any unbound bacteria by submerging the slides three times in a beaker filled with PBS. Visualize bacteria by Gram staining, as described.
- Remove excess PBS from each slide by tilting it and touching the edge onto tissue paper. Air-dry the slides for 3-5 min at RT, and fix adhered bacteria by passing each slide three times over a flame.
- Hold the slide by the edges with two fingers on a staining tray, then add 3–4 drops (200–300 µL) of 2.3% crystal violet solution. Wait 60 s, then wash the slides under a gentle stream of tap water for 3–4 s.
- Add 3–4 drops of 0.33% iodine solution onto the slides. Wait 1 min, then rinse the slide under a gentle stream of tap water. Carefully dry the slides by blotting paper.
- Add 3–4 drops of decolorizing solution containing 75% isopropyl alcohol and 25% acetone onto the slide. After 5–10 s, wash the slide under a gentle stream of tap water.
- Add 3–4 drops (200–300 µL) of basic carbolfuchsin solution. Wait 1 min, then rinse the slides under a gentle stream of tap water. Dry the slides using blotting paper.
- Visualize the bacteria under a light microscope, selecting an oil-immersion objective lens at 100X magnification18. Compare the adherence of Hif WT and mutant bacteria on the Vn- and HSA-coated glass surfaces.
- Study of Vn-dependent adherence of bacteria to epithelial cells
NOTE: This efficient adherence assay was used in our previous studies4,7.- Culture A549 cells (type II alveolar epithelial cells) in a 75-cm2 tissue culture flask with F12 medium supplemented with 10% (vol/vol) fetal calf serum (FCS) (complete medium) and 5 µg/mL gentamicin. Incubate the flask in an incubator with 5% CO2 at 37 °C for 3 days until 80% confluent (confluency can be estimated by visualizing surface coverage by the cells using inverted microscope). The following four steps describe how to prepare the epithelial cells for the assay19,20.
- Wash the cell monolayer in the flask twice with 20 mL of PBS by gentle swirling. To detach the cells from the flask surface, add 2 mL of cell detachment enzyme solution and incubate the flask for 5 min at 37 °C. Tap the flask with your palm to detach all of the cells from the plastic surface. Pipette up and down a few times to disperse cell clumps.
- Add 18 mL of F12 complete medium to the flask and transfer the entire cell suspension to a 50-mL sterile Falcon tube. Centrifuge the cell suspension for 5 min at 200 x g at RT and discard the supernatant. Resuspend the cell pellet in 10 mL of F12 complete medium.
- Remove 10 µL of the cell suspension and place it in an Eppendorf tube, then add 90 µL of Trypan blue solution. Load the sample into a hemocytometer (depth 0.1 mm) after placing the coverslip.
- Count all viable cells in areas A, B, C, and D (each field consists of 16 squares, and each square has an area of 0.0025 mm2), then calculate the average number of cells ([A+B+C+D]/4). Calculate the number of cells per milliliter using the following equation:
Viable cells/mL = average cell count × 104 × dilution factor20.
NOTE: Here, the dilution factor is 10. - Dilute the cell suspension to 5.0 x 103 cells/mL using complete medium containing 5 µg/mL gentamicin. Dispense 500 μL of cell suspension into each well of a 24-well cell culture plate. Incubate the plate at 37 °C in 5% CO2 until the cells are 90% confluent.
- Prior to bacterial infection, remove the medium from the wells and add F12 medium (without FCS), then incubate overnight at 37 °C.
- Wash the cell monolayer three times with 500 µL of PBS at RT. Place the plate on ice and add 100 µL of pre-chilled F12 medium containing 10 µg/mL of Vn. Incubate plate at 4 °C for 1 h. For control wells, add only F12 medium.
- After incubation, discard the solution by pipetting and wash the cell layer twice with 1 mL of PBS at RT. Resuspend a culture of freshly grown Hif M10 (step 1.1.1) in F12 medium (2×108 CFU/mL). Add 100 µL of this bacterial suspension to each well and incubate the plate for 2 h at 37 °C.
NOTE: Each well contains approximately 2 x 105 A549 cells. For infection, 2 x 107 bacterial CFU were added, corresponding to a multiplicity of infection of 100. - Remove the medium by pipetting and wash the A549 epithelial cells three times with PBS. Add 50 µL/well of cell detachment solution and incubate the plate for 5 min at 37 °C.
- Next, add 50 µL of F12 complete medium per well to stop the enzymatic reaction. Transfer the epithelial cells (≈100 μL [i.e., the entire volume]) from each well to a 6-mL glass tube containing four glass beads. Lyse the cells at RT by vortexing for 2 min.
- Dilute 10 µL of the cell lysate 100-fold by adding 10 µL of lysate to 990 µL of F12 medium. Plate 10 µL of the diluted sample onto a chocolate agar plate.
- Incubate the chocolate agar plate at 37 °C overnight, then count the colonies. Each colony represents one CFU.
- Culture A549 cells (type II alveolar epithelial cells) in a 75-cm2 tissue culture flask with F12 medium supplemented with 10% (vol/vol) fetal calf serum (FCS) (complete medium) and 5 µg/mL gentamicin. Incubate the flask in an incubator with 5% CO2 at 37 °C for 3 days until 80% confluent (confluency can be estimated by visualizing surface coverage by the cells using inverted microscope). The following four steps describe how to prepare the epithelial cells for the assay19,20.
3. Analysis of Vn-dependent Resistance to the Bactericidal Activity of Human Serum
- Purchase NHS from a commercial source. Prepare VDS as previously described12. Replenish the VDS with 180 nM Vn that is equivalent to Vn present in NHS.
- Prepare heat-inactivated serum (HIS) by heating NHS at 56 °C for 30 min in order to inactivate complement proteins.
NOTE: The optimal serum concentration (5%) and incubation time (15 min) for the assay described in steps 3.3–3.7 were determined empirically for Hif15; these parameters could vary for other bacterial pathogens. - Culture bacteria (in this case, Hif M10 and the mutant Hif M10Δlph) to mid-log phase (OD600 = 0.3). Pellet bacteria by centrifugation at 3,500 x g for 10 min.
- Resuspend the bacterial pellet with 1 volume of dextrose gelatin Veronal buffer (DGVB++; pH 7.3) containing 2.5% (w/v) glucose, 2 mM MgCl2, 0.15 mM CaCl2, and 0.1% (wt/vol) gelatin.
- Add 1.5 x 103 CFU of bacteria to 100 µL of DGVB++ containing 5% serum (NHS, HIS, VDS, or VDS+180 nM Vn). Incubate the sample at 37 °C for 15 min with shaking at 300 rpm.
- Remove a 10 µL aliquot from the reaction mixture at 0 min (T0 sample) and 15 min (Tt sample) and plate on chocolate agar. Incubate the plate at 37 °C overnight.
- After incubation, count the colonies appearing on the plate. Calculate the percentage of bacteria killed12 using the following equation: (CFU at Tt)/(CFU at T0) × 100.
Representative Results
Vn-binding to the surface of bacteria was determined by flow cytometry. All Hif clinical isolates tested in this study recruited Vn to the cell surface. No interaction of Vn with the cell surface was observed for the E. coli negative control strain (Figure 1A). As shown in Figure 1B, PH is a major Vn-binding protein on the surface of Hif cells. Binding of Vn by the WT Hif strain M10 caused a shift in the population, whereas Hif M10Δlph did not bind Vn and appeared similar to the control.
Protein-protein interactions between PH and Vn were estimated by ELISA and BLI. Recombinant PH was allowed to bind with human factor H, Vn, and C4BP (negative control) coated onto the wells of microtiter ELISA plates. Bound PH was estimated using an anti-His pAbs. The results clearly indicated a significant interaction between PH and Vn and factor H, in comparison to C4BP (Figure 2A). Interaction between PH and Vn were estimated in real time using BLI. Figure 2B shows PH binding response curves for the Vn-coated sensor surface. Binding affinity (in this case, Kd = 2.2 µM) was estimated by fitting the data to steady-state binding kinetics.
Bacteria lacking expression of PH on the cell surface (Hif M10Δlph) exhibited reduced adherence to Vn-coated glass surfaces in comparison to WT cells (Figure 3A). In addition, the presence of Vn on the surface of epithelial cells significantly enhanced the adherence of Hif (Figure 3B). These results indicated that the PH-Vn interaction contributed significantly to bacterial adherence.
Vn is a well-known complement inhibitor. To estimate the complement-inhibiting activity, serum-mediated killing was examined in the presence and absence of Vn. WT Hif cells exhibited higher serum resistance than M10Δlph mutant cells. No bacteria were killed in the presence of HIS (Figure 4A). Interestingly, WT Hif exhibited reduced survival in VDS, and replenishment of Vn increased bacterial survival. However, the Hif M10Δlph strain did not respond to Vn depletion because it could not recruit Vn to the cell surface (Figure 4B). These results clearly demonstrate that Vn is an important host factor that protects bacteria from complement-mediated killing (Figure 4A-B).
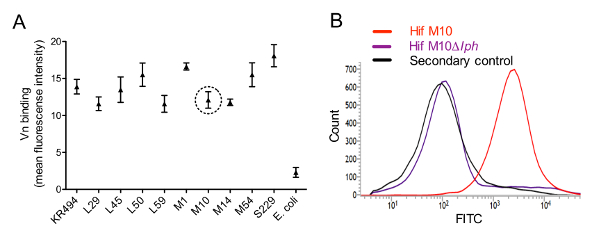
Figure 1: Haemophilus influenzae serotype f binds Vn via surface-expressed PH. (A) Flow cytometry results showing binding of Vn to cells of Hif clinical isolates. Each clinical isolate (5 x 106 CFU) was incubated with 250 nM human Vn. Bound ligand was detected using a sheep anti-Vn pAbs and FITC-conjugated donkey anti-sheep secondary antibodies. Escherichia coli BL21 (DE3) was included as a negative control. Data are presented as the mean fluorescence intensity from triplicate analyses in three separate experiments, and error bars represent SD. (B) Flow cytometry histograms demonstrating binding of Vn to the surface of WT Hif M10 and PH-deletion Hif M10Δlph mutant. Representative data from one of three separate experiments are shown. Please click here to view a larger version of this figure.
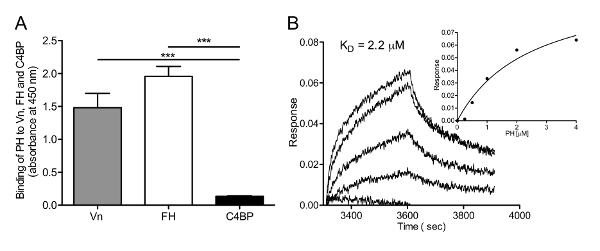
Figure 2: Recombinant PH binds to Vn. (A) ELISA results demonstrating the binding of recombinant PH to Vn, FH, and C4BP. FH was used as a positive control, whereas C4BP was used as a negative control. To analyze protein-protein interactions, Vn, FH, and C4BP were coated in equimolar (50 nM) concentrations onto the wells of microtiter plates. Next, 50 nM recombinant PH-His6x was added. Bound PH was detected using an HRP-conjugated anti-His pAbs. Data shown are the means of triplicate analyses from three independent experiments, and error bars indicate SD. ***, P≤0.001. (B) BLIanalysis of PH binding to Vn: Recombinant Vn was immobilized on AR2G sensors, and the binding kinetics of PH (0.25-4 µM) to Vn were recorded using the BLI instrument. The equilibrium affinity constant was calculated using signals obtained for each concentration at equilibrium, and the data were fitted according to steady-state kinetics. The experiment was repeated three times, and one representative dataset is shown. Please click here to view a larger version of this figure.
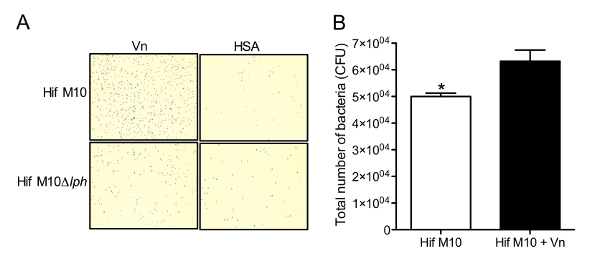
Figure 3: Interaction between PH and Vn enhances bacterial adherence to glass surfaces and epithelial cells. (A) Light microscopic images showing adherence of Hif to Vn-coated glass slides. Bacteria were visualized by Gram staining. Representative images from one of three independent experiments are shown. This panel was previously published in the Journal of Immunology7 and is reproduced here with permission from the American Association of Immunologists. (B) Adherence of WT Hif M10 cells to A549 epithelial cells in the presence and absence of Vn, means of triplicate analyses from three independent experiments are plotted, and error bars represent SD. *, p ≤0.05. Please click here to view a larger version of this figure.
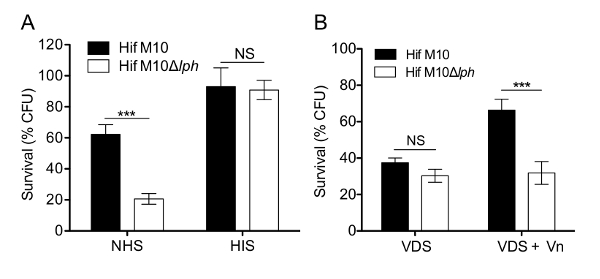
Figure 4: Vn recruited to the Hif cell surface protects bacteria from serum-mediated killing. (A) WT Hif M10 and mutant Hif M10Δlph were incubated for 15 min in 5% NHS or HIS. (B) Resistance of Hif M10 and Hif M10Δlph to bactericidal effects in 5% VDS and VDS supplemented with 250 nM purified Vn. Data represent means of triplicate analyses from three independent experiments, and error bars indicate SD. ***, p≤0.001; NS, not significant. Please click here to view a larger version of this figure.
Discussion
Bacterial pathogens recruit Vn to the cell surface and utilize this complement regulator to prevent the deposition of complement factors and completion of the membrane attack complex2. Vn also functions as a bridge molecule between bacterial surface proteins and host cell surface receptors, thus enabling pathogens to adhere to the surface of epithelial cells and subsequently mediate internalization. In this study, we describe protocols that can be used to estimate i) binding of Vn to the surface of bacterial cells, ii) protein-protein interactions and affinity constants, and iii) the functional role of Vn in providing serum resistance and enhancement of adherence to the surface of host cells.
Flow cytometry is a quantitative technique commonly used to verify that bacteria bind Vn. However, the Vn concentration must be optimized, as the Vn-binding capacity (and various receptors) can differ depending on the bacterial species. In the assay described here, 1–100 µg/mL of Vn were used to estimate linear binding and saturation parameters. Comparison of the binding patterns of WT Hif, the Hif M10Δlph mutant, and negative control E. coli cells showed a clear difference at a Vn concentration of 20 µg/mL (250 nM). Therefore, this concentration was used in all flow cytometry experiments. The use of Vn at concentrations above the saturation point could produce false-positive signals. Dilution of the primary and secondary antibodies must also be separately optimized for each pathogen. The maximum dilution of antibodies showing a good signal should be chosen. In this study, we used 2.5% BSA as a blocking agent. However, BSA is not a universal blocking reagent suitable for all pathogens. In cases in which BSA does not block nonspecific antibody interactions, other blocking reagents can be used, such as fish gelatin. Binding of Vn to the surface of bacterial pathogen cells is often mediated by multiple receptors21,22. Some receptors have high Vn-binding affinity, whereas others may exhibit low binding affinity. Thus, deletion of only one gene encoding a surface protein might not result in a decrease in Vn interactions assessed by flow cytometry. As an alternative to flow cytometry, protein binding on the bacterial cell surface can be examined using an immunofluorescence assay (IFA) or transmission electron microscopy (TEM). Although photobleaching of fluororeagent-conjugated antibodies can be problematic in both flow cytometry and IFAs, TEM involves cumbersome sample preparation, with substantial risk of introducing artefacts23,24. Flow cytometry analysis is indeed preferable to IFA and TEM due to simpler sample preparation protocols and the possibility of testing many samples within a short timeframe.
Protein-protein interactions in a cell free system were analyzed by ELISA (Figure 2A) and BLI (Figure 2B). In both of these techniques, one of the binding partners was immobilized, either on a biosensor (for BLI) or a microtiter plate (ELISA). The advantage of BLI and ELISA is that they enable analysis of protein-protein interactions in their native state, preventing nonspecific interactions outside the active binding site. The utility of BLI, however, can be limited depending on the immobilization technique used. The amine coupling procedure used here randomly forms an amide bond between any surface-exposed amine group of the protein and the surface of the biosensor. This results in immobilization of protein molecules on the surface of the sensor in various orientations. This can result in heterogeneous binding that does not fit a 1:1 binding kinetics model. This potential problem can be resolved by selecting biotin sensors and adding a streptavidin tag in the recombinant protein to be immobilized. The reliability of BLI is similar to that of surface plasmon resonance (SPR). The association and dissociation of molecules can be measured in real time using either technique. However, the BLI device does not use microfluidics, and is therefore easy to maintain. In addition, 96 interactions can be tested simultaneously in BLI, whereas only four samples at a time can be tested using SPR. There is another critical step in BLI that must be optimized. If sensors are reused, regeneration conditions should also be optimized, as that might vary for each protein-protein complex. In this study, 10 mM ethylenediaminetetraacetic acid was found suitable for sensor regeneration.
The role of Vn in bacterial adherence was tested directly using glass surfaces coated with Vn. This approach can be employed to analyze binding for any bacterial pathogen, including both Gram-negative and -positive bacteria. This is a semiquantitative technique that provides rapid assessment of a pathogen's capacity for binding to Vn. To obtain reasonable results, Vn coating must be optimized. In the assay described here, coating with Vn at 1, 2, and 5 µg/mL was examined, and 2 µg/mL was found to be the most suitable concentration for the experiments conducted in this study (Figure 3A). Excess Vn coated on the surface could create a thick layer that could be easily stripped off during sample fixing and staining. Moreover, the bacterial culture should not have large clumps of cells; this can be avoided by vortexing the culture before adding an aliquot to the slides.
Bacterial adherence to the surface of epithelial cells was examined using a plate-count technique (Figure 3A). This assay also must be carefully optimized to enable observation of the role of Vn in bacterial adherence. Vn has different binding sites for bacteria (at the C-terminus) and cell surface integrin receptors (at the N-terminal RGD binding site). Binding of Vn to integrins induces signaling and uptake of the integrin-Vn complex1. If such complexes are internalized without bacteria, results will be difficult to interpret. Therefore, Vn must be added to the epithelial cell monolayer at 4 °C. Once Vn has saturated integrin receptors at the epithelial cell surface, excess Vn must be washed off, as free Vn can block bacterial Vn-dependent integrin binding. In this assay, we estimated total bacterial counts associated with epithelial cells (i.e., the read-out comprised both surface-attached and internalized bacteria). This assay can be extended to distinguish between external and intracellular bacterial populations by treatment with gentamicin4.
The serum bactericidal assay used in the protocol described here was partially modified from the previously published procedure12,22. In the assay described here, 1.5 x 103 CFUs of Hif were incubated with 5% NHS for 15 min, 5 min longer than in the previously described assay (Figure 4A). The difference between WT Hif and the isogenic mutant devoid of PH was more evident at 15 min than at 10 min. Therefore, we selected the 15 min incubation period for this particular experiment. The bactericidal effects of serum depend on the serum concentration, time of incubation, and number of bacteria. All pathogens exhibit a notable capacity for avoiding the bactericidal activity of serum; some pathogens can be completely resistant to serum, whereas others can be moderately resistant or sensitive. The serum concentration must therefore be optimized for each specific pathogen examined. Nevertheless, the serum killing assay described here is limited to the study of gram-negative bacteria, since serum has no significant bactericidal effect on gram-positive cells1,5.
Divulgations
The authors have nothing to disclose.
Acknowledgements
This work was supported by grants from the Foundation of Anna and Edwin Berger, Lars Hierta, the O.E. and Edla Johansson Foundation, the Swedish Medical Research Council (grant number K2015-57X-03163-43-4, www.vr.se), the Cancer Foundation at the University Hospital in Malmö, the Physiographical Society (Forssman's Foundation), and the Skåne County Council's Research and Development Foundation.
Materials
| 1.5 mL thermomixter | Eppendorf | 5355 | dry block heating and cooling shaker |
| 5 mL polystyrene round-bottom tube | BD Falcon | 60819-138 | 12 × 75 mm style |
| 5% CO2 supplied incubator | Thermo Scientific | BBD6220 | |
| 6 mL polystyrene round-bottom tube | VWR | 89000-478 | 12 × 75 mm style with cap |
| 24-well plates | BD Falcon | 08-772-1H | Cell culture grade |
| 30% Hydrogen peroxide (H2O2) solution | Sigma-Aldrich | H1009-100ML | Laboratory analysis grade |
| 75 cm2 tissue culture flask | BD Falcon | BD353136 | Vented |
| 96 well black flat bottom plate | Greiner Bio-One | 655090 | Tissue culture treated µClear black plates |
| A549 Cell Line human | Sigma-Aldrich | 86012804-1VL | |
| Cell detachment enzyme (Accutase) | Sigma-Aldrich | A6964-500ML | Cell Culture Grade |
| AR2G sensors | Pall Life Science | 18-5095 | Sensor to immobilized protein by amino coupling |
| Acetone | VWR | 97064-786 | Analysis grade |
| Bovine Serum Albumins (BSA) | Sigma-Aldrich | A2058 | Suitable for cell culture |
| Bibulous paper | VWR | 28511-007 | |
| Bio-layer interferometer | Pall Life Science | FB-50258 | Bilayer interferometry measuring equipment |
| Crystal violet solution | Sigma-Aldrich | HT90132-1L | |
| C4BP (C4b binding protein) | Complement Technology, Inc. | A109 | Bought as Frozen liquid form |
| Calcium chloride (CaCl2) | Sigma-Aldrich | C5670-500G | |
| Carbol-fuchsin solution | Sigma-Aldrich | HT8018-250ML | |
| Citric acid | Sigma-Aldrich | 251275-500G | American Chemical Society (ACS) grade |
| Decolorizing solution | Sigma-Aldrich | 75482-250ML-F | |
| E. coli host (E. Coli BL21) | Novagen | 69450-3 | Protein expression host |
| F12 medium | Sigma-Aldrich | D6421 | Cell Culture Grade |
| Flow cytometer | BD Biosciences | 651154 | Cell analysis grade for research applications |
| Fetal Calf Serum (FCS) | Sigma-Aldrich | 12003C | Suitable for cell culture |
| Normal human serum (NHS) | Complement Technology, Inc. | NHS | Pooled human serum |
| FITC-conjugated donkey anti-sheep antibodies | AbD Serotec | STAR88F | Polyclonal |
| Gentamicin | Sigma-Aldrich | G1397 | Cell culture grade |
| Glucose | Sigma-Aldrich | G8270-1KG | |
| Gelatin | Sigma-Aldrich | G9391 | Suitable for cell culture |
| Hemocytometer | Marienfeld | 640210 | |
| HRP-conjugated anti-His tag antibodies | Abcam | ab1269 | Polyclonal |
| Human factor H | Complement Technology, Inc. | A137 | Bought as Frozen liquid form |
| C4BP | Complement Technology, Inc. | A109 | Frozen solution |
| Human serum albumin | Sigma-Aldrich | A1653-10G | lyophilized powder |
| Histidine affinity resin column (HisTrap HP) | GE Health Care Life Science | 17-5247-01 | Columns prepacked with Ni Sepharose |
| His-tagged PH | Recombinantly expressed and purified in our lab | ||
| Iodine solution | Sigma-Aldrich | HT902-8FOZ | |
| Methanol | VWR | BDH1135-1LP | Analysis grade |
| Microscope | Olympus | IX73 | Inverted microscope |
| Microscope slides | Sigma-Aldrich | S8902 | plain, size 25 mm × 75 mm |
| Magnesium chloride (MgCl2) | Sigma-Aldrich | M8266-1KG | |
| Plasmid containg C terminal 6x His-tag on the backbone (pET26(b)) | Novagen | 69862-3 | DNA vector |
| Polysorb microtitre plates | Sigma-Aldrich | M9410 | For ELISA |
| Potassium hydroxide (KOH) | Sigma-Aldrich | 6009 | American Chemical Society (ACS) grade |
| Sheep anti-human Vn antibodies | AbD Serotec | AHP396 | Polyclonal |
| Shaker | Stuart Scientific | STR6 | Platform shaker |
| Tissue culture flask | BD Falcon | 3175167 | 75 cm2 |
| Thermomixer | Sigma-Aldrich | T3317 | Dry block heating and cooling shaker |
| Tetramethylbenzidine | Sigma-Aldrich | 860336-100MG | ELISA grade |
| Vitronectin (Vn) from human plasma | Sigma-Aldrich | V8379-50UG | cell culture grade |
References
- Singh, B., Su, Y. C., Riesbeck, K. Vitronectin in bacterial pathogenesis: a host protein used in complement escape and cellular invasion. Mol. Microbiol. 78 (3), 545-560 (2010).
- Hallstrom, T., et al. Conserved Patterns of Microbial Immune Escape: Pathogenic Microbes of Diverse Origin Target the Human Terminal Complement Inhibitor Vitronectin via a Single Common Motif. PloS one. 11 (1), 0147709 (2016).
- Su, Y. C., Hallstrom, B. M., Bernhard, S., Singh, B., Riesbeck, K. Impact of sequence diversity in the Moraxella catarrhalis. UspA2/UspA2H head domain on vitronectin binding and antigenic variation. Micro. Infect. 15 (5), 375-387 (2013).
- Singh, B., et al. A fine-tuned interaction between trimeric autotransporter haemophilus surface fibrils and vitronectin leads to serum resistance and adherence to respiratory epithelial cells. Infect. Immun. 82 (6), 2378-2389 (2014).
- Kohler, S., et al. Binding of vitronectin and Factor H to Hic contributes to immune evasion of Streptococcus pneumoniae. serotype 3. Thromb. haemostasis. 113 (1), 125-142 (2015).
- Onelli, E., Citterio, S., O’Connor, J. E., Levi, M., Sgorbati, S. Flow cytometry, sorting and immunocharacterization with proliferating cell nuclear antigen of cycling and non-cycling cells in synchronized pea root tips. Planta. 202 (2), 188-195 (1997).
- Al-Jubair, T., et al. Haemophilus influenzae Type f Hijacks Vitronectin Using Protein H To Resist Host Innate Immunity and Adhere to Pulmonary Epithelial Cells. J. Immunol. 195 (12), 5688-5695 (2015).
- Martinez-Martin, N. Technologies for Proteome-Wide Discovery of Extracellular Host-Pathogen Interactions. J. Immunol. Res. 2017, 2197615 (2017).
- Boleij, A., Laarakkers, C. M., Gloerich, J., Swinkels, D. W., Tjalsma, H. Surface-affinity profiling to identify host-pathogen interactions. Infect. Immun. 79 (12), 4777-4783 (2011).
- Abdiche, Y., Malashock, D., Pinkerton, A., Pons, J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal. Biochem. 377 (2), 209-217 (2008).
- Sultana, A., Lee, J. E. Measuring protein-protein and protein-nucleic Acid interactions by biolayer interferometry. Cur. Prot. Prot. Sc. 79, 11-26 (2015).
- Su, Y. C., et al. Haemophilus influenzae acquires vitronectin via the ubiquitous Protein F to subvert host innate immunity. Mol. Microbiol. 87 (6), 1245-1266 (2013).
- Ronander, E., et al. Nontypeable Haemophilus influenzae adhesin protein E: characterization and biological activity. J. Infect. Dis. 199 (4), 522-531 (2009).
- Sezonov, G., Joseleau-Petit, D., D’Ari, R. Escherichia coli physiology in Luria-Bertani broth. J. Bact. 189 (23), 8746-8749 (2007).
- Fleury, C., et al. Identification of a Haemophilus influenzae factor H-Binding lipoprotein involved in serum resistance. J. Imunol. 192 (12), 5913-5923 (2014).
- Abdiche, Y., Malashock, D., Pinkerton, A., Pons, J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal. Biochem. 377 (2), 209-217 (2008).
- Rich, R. L., Myszka, D. G. Higher-throughput, label-free, real-time molecular interaction analysis. Anal. Biochem. 361 (1), 1-6 (2007).
- McNamara, G., Difilippantonio, M. J., Ried, T. Microscopy and image analysis. Current protoc. Hum. Genet. , 4 (2005).
- Lieber, M., Smith, B., Szakal, A., Nelson-Rees, W., Todaro, G. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. International J. Canc. 17 (1), 62-70 (1976).
- Foster, K. A., Oster, C. G., Mayer, M. M., Avery, M. L., Audus, K. L. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp. Cell Res. 243 (2), 359-366 (1998).
- Su, Y. C., et al. Haemophilus influenzae P4 Interacts With Extracellular Matrix Proteins Promoting Adhesion and Serum Resistance. J. Infect. Dis. 213 (2), 314-323 (2016).
- Singh, B., Al-Jubair, T., Morgelin, M., Thunnissen, M. M., Riesbeck, K. The unique structure of Haemophilus influenzae protein E reveals multiple binding sites for host factors. Infect. Immun. 81 (3), 801-814 (2013).
- Vellaiswamy, M., Campagna, B., Raoult, D. Transmission electron microscopy as a tool for exploring bacterial proteins: model of RickA in Rickettsia conorii. New Microbiolog. 34 (2), 209-218 (2011).
- Pinne, M., Haake, D. Immuno-fluorescence assay of leptospiral surface-exposed proteins. J. Vis. Exp. JoVE. (53), (2011).
