The care and treatment of animals in this procedure were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee at the University of Colorado.
1. Preparing Solutions
- Prepare and store all solutions at appropriate temperatures, as shown in Table 1.
2. Coating Wells and Slides
NOTE: Neurons do not adhere well to plastic or glass surfaces.
- One day prior to the isolation of the neurons, coat the wells of a sterile 24-well culture plate with 0.5 mL of Poly-D-Lysine (PDL; Table 2) and leave it in a laminar flow hood O/N.
NOTE: The preferred method is to use 24-well plates and to coat only the center wells, as evaporation from the peripheral wells tends to be more accelerated and to lead to inconsistent results. Additionally, glass slides can be placed inside the wells prior to coating. The neurons will attach to the coated glass slides and can be removed later for further microscopic imaging. - On the day of isolation, remove the PDL from the wells. Wash with sterile water for a few minutes before removing any remaining water and allowing it to dry for 1 h (see below).
- After drying, coat the slides with laminin (Table 2).
NOTE: Approximately 10 μL of laminin (10 µg/µL) is enough to coat each well of a sterile 24-well plate. This is mixed with 340 μL of medium (total volume: 350 μL) in order to cover the entire well. Let this sit for 2 h at RT in a culture hood and then aspirate prior to coating the cells.
3. Harvesting Spinal Cords
NOTE: All instruments should be autoclaved (135 °C and 30 psi for 4 x 7 min cycles) for sterility.
- Euthanize 1-3 day-old C57BL/6 mouse pups in a chamber with isoflurane. Wait 30 s after the cessation of movement and pinch the leg to confirm a lack of response.
- Separate the head from the body using scissors, with pup in the prone position.
- Stabilize the hind legs or tail and arms on the procedure table, with dorsal side facing the user.
- Cut the skin off using curved iris scissors.
- Cut the spinal cord from the lumbar region just above the hips and proceed to cut both sides of the thorax to separate it from the body.
NOTE: This requires the careful dissection of the spinal cord from visceral organs to avoid inadvertent damage to other organs (Figure 1a). - Wash sequentially for 10 s in 3 x 10 cm Petri dishes containing 5 mL of 0.2 μm filter-sterilized Phosphate-Buffered Saline (PBS) to remove excess tissue.
- Insert a 22 G needle and syringe filled with 5 mL of filter-sterilized PBS into the caudal end of the spinal column and flush cranially, allowing the cord to exit into a fourth Petri dish (Figure 1b).
- Collect the spinal cord in a 15 mL tube with 5 mL of HABG (Table 1) on ice. Use care to avoid crushing the spinal cord.
- Repeat steps 3.1-3.8 for each pup in the litter.
NOTE: Ideally, this process should take less than 30 min per spinal cord to ensure healthy neuron isolation.
4. Isolating Neurons
NOTE: The following step should be performed in a laminar flow hood. Familiarity with basic sterile technique is expected.
- Tissue Mincing
- Take the tube containing the spinal cords and shake lightly to suspend the tissue.
- Pour tissue from the tube into a 60 mm glass Petri dish and dice with a razor blade to create fine pieces ~0.5 mm in size.
- Transfer the tissue with a wide-bore pipette into a 15 mL tube containing 5 mL of HABG.
- Place it in a 30 °C water bath for 30 min to allow the cells to equilibrate at this temperature. Keep the cells on a shaker just enough to allow them to be suspended in the fluid.
NOTE: This step is done to avoid shocking the cells upon transfer from ice to the digestion medium. Keeping the cells at 30 °C helps to decrease cell death associated with an otherwise increased metabolism at 37 °C.
- Prepare the digestion medium (Table 1).
- Prepare the density gradient (Table 1).
- Prepare each of the 4 layers in 4 separate 15 mL tubes, as outlined in Table 1.
- Add 1 mL from each layer into a new 15 mL tube. Start with layer 1 at the bottom and sequentially add until reaching layer 4 at the top. Avoid disturbing the layers while adding.
- Wash the PDL-coated plates from step 2.2. Wash with sterile water for a few minutes before removing any remaining water and allowing them to dry for 1 h.
- Transfer the tissue to digestion medium.
- Remove the tissue-containing tube from the shaker water bath at 30 °C and allow it to settle for a few minutes.
- Remove the digestion medium tube from the 37 °C water bath and aspirate it into a leur-lock syringe.
- Aspirate off the excess HABG from the tissue-containing tube.
- Use a leur-lock 0.2 μm filter on the syringe to add digestion medium to the tissue-containing tube.
- Place the tube in a 30 °C water bath for 30 min. Keep the cells shaking just enough to allow them to be suspended in the fluid.
NOTE: It is important not to keep the cells in the digestion medium for too long or to let the temperature get too high, which could lead to excessive digestion and result in the tissue becoming suspended in a gelatinous mixture.
- During this period, coat the laminin as in step 2.3.
- Perform trituration (i.e. separating the cells from the tissue).
- Remove the tube from the shaking 30 °C water bath and allow it to settle for a few min.
- Aspirate excess digestion medium.
- Suspend the tissue in 2 mL of HABG.
- Using a narrow-bore pipette, triturate 10x for 45 s.
NOTE: This is probably the single most crucial step and can significantly decrease the yield if not done properly.- Aspirate the tissue into the pipette and immediately empty the contents back.
- Avoid introducing air, as it will significantly decrease the viable yield.
NOTE: The ideal pipette is a 9" glass pipette. The tip of the pipette should be fire polished to smooth out rough surfaces. It should then be siliconized by placement in a 1:20 solution of dichlorodimethylsilane (DMDCS) in chloroform and left O/N. The pipette should then be removed and allowed to air dry. Subsequently, it should be autoclaved for sterilization.
Caution: DMDCS and chloroform are highly flammable, and siliconizing should be carried out in a fume hood.
- Aspirate the top 2 mL of supernatant and place it into a new 15 mL tube labeled "collection."
- Repeat steps 4.7.3-4.7.5 two additional times (the cell collection tube should have 6 mL by the end).
- Slowly transfer the collection tube contents into the gradient tube prepared in step 4.2, avoiding the disruption of the gradient.
- At this point, remove the previously prepared neurobasal medium (Table 1) from the refrigerator and allow it to warm in a 37 °C bath.
- Purify the neurons.
- Centrifuge the gradient tube for 15 min at 800 x g and 22 °C.
- Collect the desired layer(s) with a pipette (Figure 2) and place in a new 15 mL tube. For the highest-purity neuron isolation (i.e. >90%), collect layer 3. For more yield with less purity (i.e. >70-80%), collect layers 2 & 3.
- Dilute out the density gradient by adding 5 mL of HABG to the newly collected layers.
- Centrifuge at 200 x g for 2 min at 22 °C.
- Discard the supernatant, re-suspend in 5 mL of HABG, and flick the pellet to suspend the cells.
- Centrifuge at for 2 min 200 x g at 22 °C.
- Discard the supernatant, resuspend in 3 mL of neurobasal medium, and flick the pellet to resuspend the cells.
- Count the cells.
- Take 10 μL of the solution, now with cells in neurobasal medium, and mix with 10 μL of Trypan blue.
- Place 10 μL of this mixture in a glass counting chamber.
- Using a standard glass counting chamber, count the number of cells in each of the four 4 x 4 quadrants. Add all of the cells counted (n), multiply by 2 (dilution factor), divide by 4 (number of quadrants counted), multiply by 3 (volume of neurobasal medium), and multiply by 104 to obtain the concentration of cells in cells/mL.
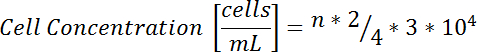
- Seed the cells on culture plates
- Dilute the cell suspension to 300,000 cells in 1 mL of neurobasal medium.
NOTE: Based on the concentration of cells obtained in the above steps, additional neurobasal medium is added to obtain a final concentration of 3*105 cells/mL. The equation is C1V1=C2V2, where C1 is the initial concentration of cells obtained from the harvest; V1 is 3 mL; and C2 is 3*105 cells/mL, as discussed above. The equation is solved for V2. Add the volume of neurobasal medium necessary to make the total volume of cells in solution equal to V2. - Shake gently to distribute the cells in solution and add 1 mL to each well in the coated 24-well plates.
- Dilute the cell suspension to 300,000 cells in 1 mL of neurobasal medium.
Using this technique, a single litter (4-10 pups) allows for the isolation of 1-2.5 106 neurons suitable for seeding onto culture plates. Typically, 4-8 wells are seeded at the concentration mentioned above (i.e. 300,000 cells/mL). Figure 3 demonstrates the appearance of neurons at this concentration after a week in culture at low- (a) and high- (b) magnification light microscopy. However, we have also been able to culture cells at concentrations as high as 500,000 cells/well and as low as 100,000 cells/well. Using the higher concentrations requires more medium and careful attention to environmental conditions, as nutrients can be depleted quickly, leading to a toxic acidic environment for the cells. With lower concentrations, cells tend not to reach full maturity, and an overgrowth of supporting cells (i.e. oligodendrocytes, microglia, and astrocytes) is observed.
The cells will typically start adhering to the surface within the first couple of hours (Figure 4a). Axons will begin to sprout within the first 24-48 h (Figure 4b-4c). Connections between various neurons in culture typically reach maturity at 7 days (Figure 4d), at which point experiments are typically carried out on the neurons.
Neurons are identifiable under light microscopy, as they have distinct axonal projections. Our isolations using layer 3 (Figure 2) yields ~80-90% neurons in culture. This was confirmed using the immunofluorescent staining of neuron-specific markers. In Figure 5c, the neuron cytoskeletal protein Microtubule-Associated Protein 2 (MAP2) is stained, showing outlines and axonal projections of the neurons after a week in culture. Similarly, in Figure 6c, the neuron-specific nuclear marker NeuN is stained, showing neuronal nuclei. These neuronal markers are combined with nucleus (DAPI, Figure 5a, 6a) and cytoplasm (GFP, Figure 5b, 6b) imaging, and the resulting images are merged (Figure 5d, 6d), detailing the relative abundance of neurons to other cells in culture. When using layers 2 and 3, the yield of neurons is slightly higher; however, there tend to be fewer neurons in the pure solution (~70%).

Figure 1: Spinal Column Dissected and Spinal Cord Released from a 3 day-old Neonatal Mouse. The arrow in (a) points towards the caudal end of the spine, where a needle is inserted to release the spinal cord. A released spinal cord (b) is shown submerged in PBS. Please click here to view a larger version of this figure.
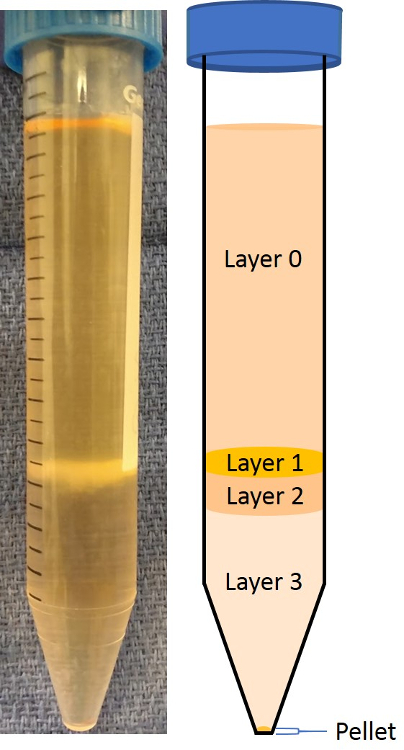
Figure 2: Density Gradient, with Cells Added-on after Centrifugation. The layers are outlined and better visualized on the cartoon depiction. The most superficial layer (0) is generally debris from the spinal cord tissue. Layer 1 is rich in oligodendrocytes and astrocytes. Layers 2 and 3 contain the majority of neurons. While layer 3 contains neurons with the highest purity, some supporting cells (i.e., astrocytes and oligodendrocytes) are found in layer 2. The pellet at the bottom mainly contains microglial cells. Please click here to view a larger version of this figure.
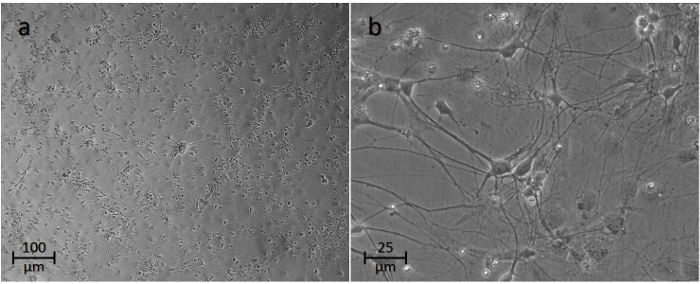
Figure 3: Light Microscopy of Neuronal Cell Cultures after 7 days of Isolation. At 10X magnification (a), axonal connections among various conglomerates of neurons can be seen. At 40X magnification (b), the neurons can be visualized more closely with their axonal projections. Please click here to view a larger version of this figure.
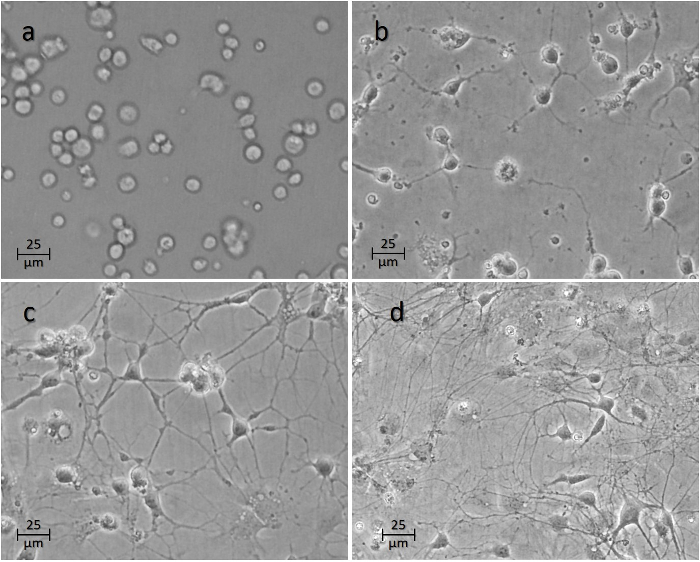
Figure 4: Time Course of the Neurons after Seeding onto the Wells. Image (a) shows the cells 1 h, (b) 24 h, (c) 48 h, and (d) 1 week after seeding. Please click here to view a larger version of this figure.
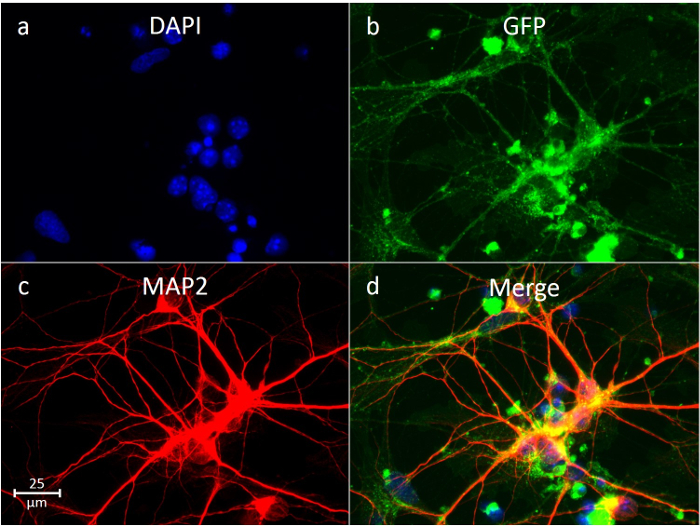
Figure 5: Immunofluorescent Stain of Neurons using Neuronal Cytoskeletal Marker MAP2. Nuclear staining (DAPI) is shown in blue (a), with cytoplasmic staining (GFP) in green (b) and neuron cytoskeletal protein (MAP2) in red (c). The merged images (d) are combined, showing the relative abundance of neurons. Please click here to view a larger version of this figure.
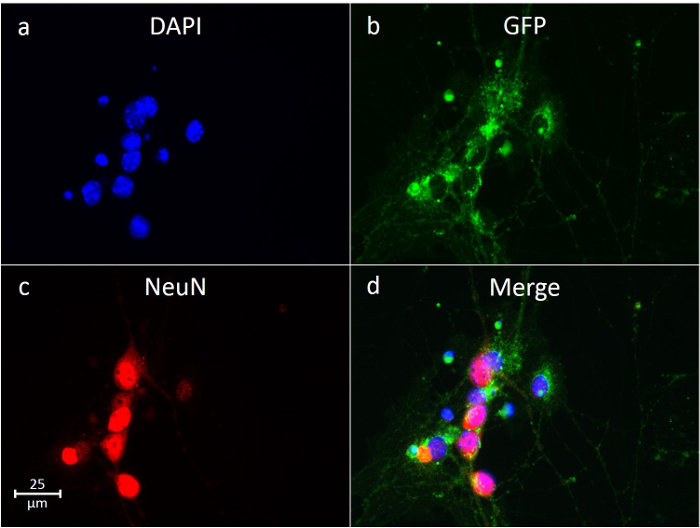
Figure 6: Immunofluorescent Stain of Neurons using Neuronal Nuclear Marker NeuN. Nuclear staining (DAPI) is shown in blue (a), with cytoplasmic staining (GFP) in green (b) and neuronal nuclei (NeuN) in red (c). The merged images (d) are combined, showing the relative abundance of neurons. Please click here to view a larger version of this figure.
| Media | Storage | Ingredients | Preparation | ||
| HABG | 4 °C/24h | 100 mL | – Thaw B27 in 4 °C fridge O/N – Filter sterilize (0.22 µm) |
||
| Hibernate A | 97.8 mL | ||||
| B27 [2%] | 2 mL | ||||
| GlutaMAX [0.5 mM] | 0.25 mL | ||||
| Neurobasal Media | 4 °C/1 week | 100 mL | – Thaw B27 in 4 °C fridge O/N – Filter sterilize (0.22 µm) - Aliquot 50 mL portions into 50 mL tubes (store at 4 °C) |
||
| Neurobasal A | 97 mL | ||||
| B27 [2%] | 2 mL | ||||
| GlutaMAX [0.5 mM] | 0.25 mL | ||||
| Pen/Strep [1%] | 1 mL | ||||
| Digestion Media | Day of Isolation | 10 mL | – Shake vigorously – Let it sit in 37°C bath for 30 min (ideally prepared 30 min prior to use) – Filter sterilize (0.22 µm) |
||
| HA-Ca | 10 mL | ||||
| Papain | 25 mg | ||||
| GlutaMAX [0.5 mM] | 0.025 mL | ||||
| Density Gradient | Day of Isolation | 1 Gradient | |||
| Layer | OptiPrep | HABG | |||
| (0.13 mL) | (5.29 mL) | ||||
| 1 Bottom | 0.26 mL | 1.24 mL | |||
| 2 | 0.19 mL | 1.31 mL | |||
| 3 | 0.15 mL | 1.35 mL | |||
| 4 Top | 0.11 mL | 1.39 mL | |||
Table 1: Preparation of the Media and Solutions used in this Protocol.
| Coating Substrate | Storage | Ingredients | Preparation | |
| PDL | -20 °C Freeze-thaw once |
Poly-D-Lysine Hydrobromide | 5 mg | – Aliquot 4 mL into 15 mL tubes and store immediately in -20 °C – 0.5 mL/well (24-well plate) |
| Sterile Water | 50 mL | |||
| Laminin | Day of coating | Laminin (1 mg/mL) | 80 µL | – Enough for 8 wells in a 24-well plate – 0.35 mL/well (24-well plate) |
| Neurobasal Medium | 2.72 mL | |||
Table 2: Preparation of the Coating Substrates used to Coat the Wells upon Which the Neurons are Cultured.
