To demonstrate the efficiency of the microinjection protocol, gRNAs designed to target the channel catfish toll/interleukin 1 receptor domain-containing adapter molecule (TICAM1) gene and rhamnose binding lectin (RBL) gene were microinjected.
TICAM 1
DNA sequencing of vectors from individual colonies from pooled, cloned PCR products revealed the indel mutations induced in TICAM 1 gene. The mutation rate was 79% in the 24 individuals analyzed. Examples of mutations are illustrated in Figure 4 and Figure 5. Effects of indel mutations in the TICAM1 gene coding sequence of channel catfish on predicted protein length and sequence compared to the wild type sequence are illustrated in Table 1. Deletion mutations resulted in removal of a few to several amino acids from the predicted protein, leading to changing the downstream reading frame and prematurely terminating translation (Table 1).
In most cases, more than 80% of the predicted protein sequence was truncated due to a premature stop codon. In these cases, the predicted polypeptide length ranged from 91 to 106 amino acid residues (wild type TICAM 1 has 520 amino acid residues). Mutations with extremely truncated protein are expected to produce a non-functional protein. In the case of the 87 bp deletion mutation (Table 2, Figure 4), 29 amino acids were removed from the protein without changing the reading frame. The functional consequences of such mutations need to be experimentally evaluated.
RBL
DNA sequencing of vectors from individual colonies from pooled, cloned PCR products confirmed the presence of indel mutations (Figure 6). The mutation rate was 88% in the 40 individuals analyzed. Deletions ranged from 5 bp to 183 bp, while up to 20 bp were inserted. Interestingly, the 183 bp deletions completely removed intron 1, exon 2, and 19 bp from intron 2. Theoretically, this should influence the splicing of the RBL gene since the splice sites have been mutated.
Indel mutations had variable effects on the predicted protein sequence when compared to the wild type protein sequence (308 amino acid residues) (Table 2). More than 70% of indels resulted in a predicted truncated protein that is 10% or less than the length of the wild type protein. In some indels (mutation number 4, 5, and 6), the predicted polypeptide was less than 10 amino acids. Only 2 cases (number 2 and 3) resulted in deletion of 2 and 4 amino acids in the predicted protein without changing the reading frame which may or may not affect the protein function.
Bi-allelic mutations were induced with two other gRNAs (Elaswad et al., unpublished) where both chromosomes were mutated in some embryos and no wild type alleles were detected. With gel electrophoresis of PCR products, these embryos had only one band that was shorter than the wild type band (more than 200 bp deletion). DNA sequencing of cloned PCR products confirmed the presence of only one mutant allele in all vectors sequenced from one embryo (homozygous biallelic mutation) while two mutant alleles occurred in other embryos (heterozygous biallelic mutation). In some other embryos that were mosaic for the mutation, both mutant (up to 4 alleles) and wild type alleles were detected by DNA sequencing of cloned PCR products.
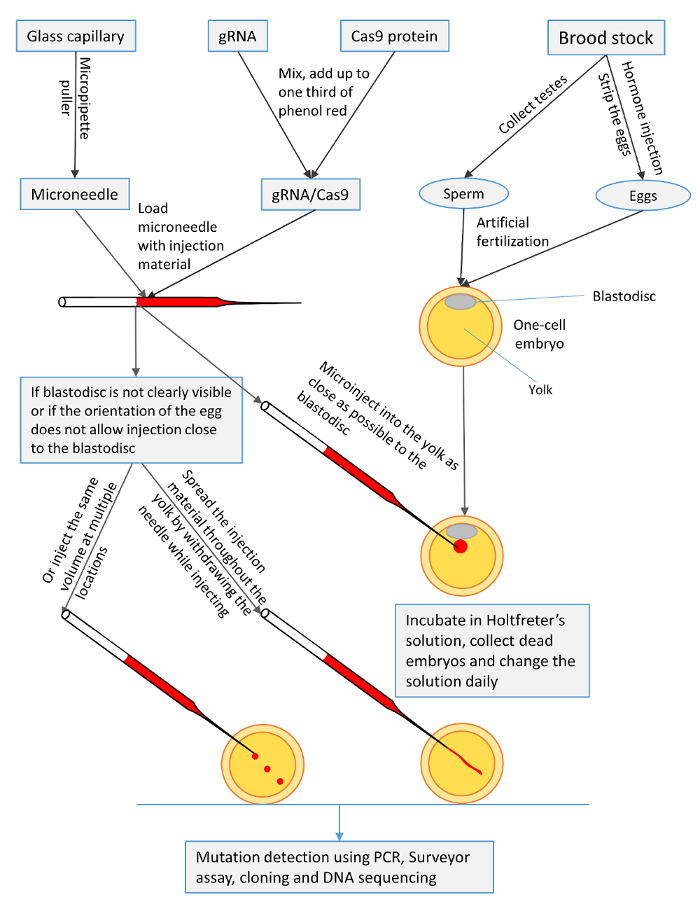
Figure 1: A summary of the microinjection procedures for one-cell channel catfish (Ictalurus punctatus) embryos with gRNA/Cas9 protein to knock out toll/interleukin 1 receptor domain-containing adapter molecule (TICAM1) gene and rhamnose binding lectin (RBL) gene. Please click here to view a larger version of this figure.
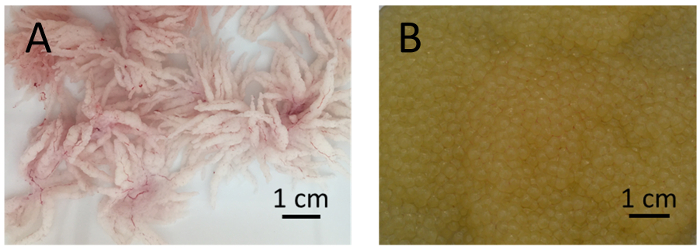
Figure 2: Testes and eggs of channel catfish, Ictalurus punctatus. (A) Well-developed testes of channel catfish males are white with many villiform projections. (B) Good eggs from channel catfish females are golden-yellow in color with minimal or no blood clots. Please click here to view a larger version of this figure.

Figure 3: Microneedle pulling, loading and microinjection of one-cell channel catfish, Ictalurus punctatus, embryos.(A) Needle pulling with a vertical needle puller. Needle structure and diameter are affected by the heat of the heater filament and the solenoid force. (B) Microneedle loading with the CRISPR/Cas9 mix using a microloader tip. (C) Arrangement of channel catfish embryos in a 100-mm Petri dish containing Holtfreter's solution. Microneedle is lowered until it touches and pierces the egg. (D) Magnified section of panel C showing the process of piercing the egg. Note the needle tip is pushing the chorionic membrane inward but has not penetrated it yet. Injected embryos have the red injection material inside. Please click here to view a larger version of this figure.
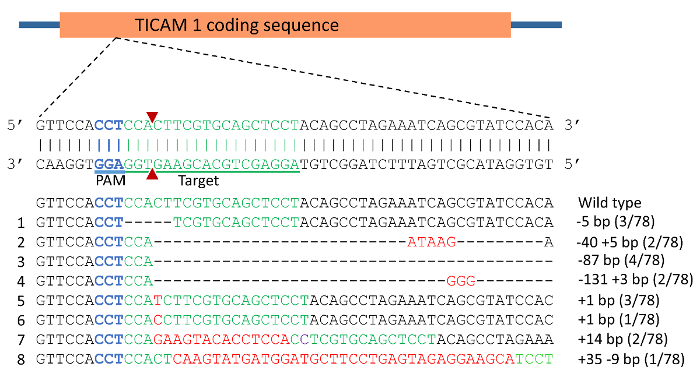
Figure 4: CRISPR/Cas9 induced mutations in toll/interleukin 1 receptor domain-containing adapter molecule (TICAM1) gene coding sequence of channel catfish, Ictalurus punctatus. Blue sequence represents the protospacer adjacent motif (PAM) while the green sequence represents the target for gRNA. Double strand break induced by Cas9 protein was expected to occur at the site of the 2 red triangles. Mutations are assigned numbers 1 through 8. Deletion mutations are represented by a dashed line in which each dash corresponds to a nucleotide that has been deleted. Red sequences are insertions while purple represents substitution mutation. Fractions of 78 represent the number of mutated alleles in 78 sequencing reactions (e.g., 3/78 means that an allele was detected in 3 sequencing reactions from a total of 78 reactions). Please click here to view a larger version of this figure.
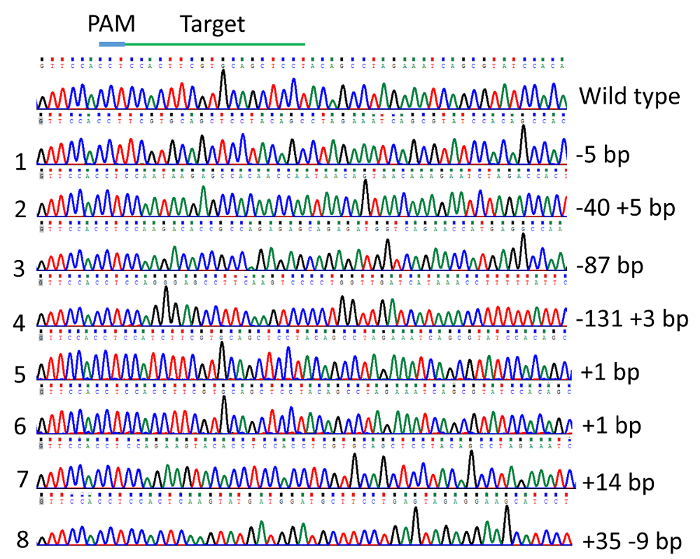
Figure 5: Indel mutations in the channel catfish, Ictalurus punctatus, toll/interleukin 1 receptor domain-containing adapter molecule (TICAM1) gene coding sequence induced by microinjection of gRNA/Cas9 protein into channel catfish one-cell embryos as revealed by DNA sequencing chromatogram. Please click here to view a larger version of this figure.
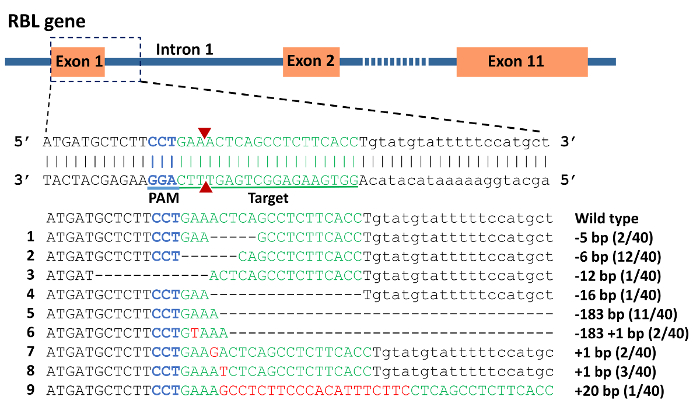
Figure 6: CRISPR/Cas9 induced mutations in rhamnose binding lectin (RBL) gene of channel catfish, Ictalurus punctatus. Exonic sequences are uppercase while intronic sequences are lowercase letters. Blue sequence represents the protospacer adjacent motif (PAM) while the green sequence represents the target for gRNA. A double strand break induced by Cas9 protein was expected to occur at the site of the 2 red triangles. Mutations are assigned numbers 1 through 9. Deletion mutations are represented by a dashed line in which each dash corresponds to a nucleotide that has been deleted. Red sequences are insertions. Fractions of 40 represent the number of mutated alleles in 40 sequencing reactions e.g. 2/40 means that an allele was detected in 2 sequencing reactions from a total of 40 reactions. Please click here to view a larger version of this figure.
| Mutation | Insertion | Deletion | Net change (Δ) | Protein length (amino acid) | Frameshift changed | Premature stop codon | Predicted protein sequence | |||
| WT | 0 | 0 | 0 | 520 | No | No | MAEETELVDEKKPTELCVNRNTVVNSPLERSISG SRSTENISGEHTAAECLIMGASLQPRRESTEAFK VQKEPPKRDDSYPSSLRSTSTSCSSYSLEISVST ATTNNSNKESRPLPLKTPPESRDGQNHEAKPSS PLVDHKPFYSVTKSYKSSDPTPLQERAQLEKFKF RQYPDKHDTSEIVTPKSPNIESGLEKTFLSIPSGG GKVGAQPVSTSKPPEASSTQEVGKHDSFSTEKQ ASQEEEDMFYAFVILHAEEDSEEAVRLKSRLESIS STIGATFSEDFAVPGQSTFRSVEDAIENSAYVMLL LTPNFNTHLNETNADSALMNSIEKPHKHNTVIPL LPRANGLTRNQMPFILRTKNPLVETRDRDTFEKM AKKVLDLRNIQRQKSMWTEAQLVKKQREKQQW LQEKKRYCKDFIQESPRVRELEEQIQQLKMQQQ HLQPPYAQQTNSHQGFPGRPQSSGPMPFRSPS PMPSYYSGNMWPQLPSNIHIQNAKCIMIGNNSTM TVGGGVDSGDEDNF (GenBank: NP_001187154.1 |
|||
| 1 | 0 | 5 | -5 | 104 | Yes | Yes | MAEETELVDEKKPTELCVNRNTVVNSPLERSISG SRSTENISGEHTAAECLIMGASLQPRRESTEAFK VQKEPPKRDDSYPSSLRSTFVQLLQPRNQRIHS HNQ |
|||
| 2 | 5 | 40 | -35 | 94 | Yes | Yes | MAEETELVDEKKPTELCVNRNTVVNSPLERSIS GSRSTENISGEHTAAECLIMGASLQPRRESTEAF KVQKEPPKRDDSYPSSLRSTSNKSHNQ |
|||
| 3 | 0 | 87 | -87 | 491 | No | Yes | MAEETELVDEKKPTELCVNRNTVVNSPLERSISG SRSTENISGEHTAAECLIMGASLQPRRESTEAFK VQKEPPKRDDSYPSSLRSTSKTPPESRDGQNHE AKPSSPLVDHKPFYSVTKSYKSSDPTPLQERAQ LEKFKFRQYPDKHDTSEIVTPKSPNIESGLEKTFL SIPSGGGKVGAQPVSTSKPPEASSTQEVGKHDSF STEKQASQEEEDMFYAFVILHAEEDSEEAVRLKS RLESISSTIGATFSEDFAVPGQSTFRSVEDAIENSA YVMLLLTPNFNTHLNETNADSALMNSIEKPHKHN TVIPLLPRANGLTRNQMPFILRTKNPLVETRDRDT FEKMAKKVLDLRNIQRQKSMWTEAQLVKKQREK QQWLQEKKRYCKDFIQESPRVRELEEQIQQLKMQ QQHLQPPYAQQTNSHQGFPGRPQSSGPMPFRSP SPMPSYYSGNMWPQLPSNIHIQNAKCIMIGNNST MTVGGGVDSGDEDNF |
|||
| 4 | 3 | 131 | -128 | 95 | Yes | Yes | MAEETELVDEKKPTELCVNRNTVVNSPLERSISG SRSTENISGEHTAAECLIMGASLQPRRESTEAFK VQKEPPKRDDSYPSSLRSTSRAFKSPG |
|||
| 5 | 1 | 0 | 1 | 106 | Yes | Yes | MAEETELVDEKKPTELCVNRNTVVNSPLERSISG SRSTENISGEHTAAECLIMGASLQPRRESTEAFK VQKEPPKRDDSYPSSLRSTSIFVQLLQPRNQRIH SHNQ |
|||
| 6 | 1 | 0 | 1 | 106 | Yes | Yes | MAEETELVDEKKPTELCVNRNTVVNSPLERSISG SRSTENISGEHTAAECLIMGASLQPRRESTEAFK VQKEPPKRDDSYPSSLRSTSTFVQLLQPRNQRIH SHNQ |
|||
| 7 | 14 | 0 | 14 | 100 | Yes | Yes | MAEETELVDEKKPTELCVNRNTVVNSPLERSISG SRSTENISGEHTAAECLIMGASLQPRRESTEAFK VQKEPPKRDDSYPSSLRSTSRSTPPPRAAPTA |
|||
| 8 | 35 | 9 | 26 | 91 | Yes | Yes | MAEETELVDEKKPTELCVNRNTVVNSPLERSISG SRSTENISGEHTAAECLIMGASLQPRRESTEAFK VQKEPPKRDDSYPSSLRSTSTQV |
|||
Table 1: Effects of indel mutations in the toll/interleukin 1 receptor domain-containing adapter molecule (TICAM1) gene coding sequence of channel catfish, Ictalurus punctatus, on predicted protein length (amino acids) and sequence compared to the wild type sequence (WT).
| Mutation | Insertion | Deletion | Net change (Δ) | Protein length (amino acid) | Frameshift changed | Premature stop codon | Predicted protein sequence | |||
| WT | 0 | 0 | 0 | 308 | No | No | MMLFLKLSLFTLIISAPGLMVSGENMITCYSDVQR LTCETGLIKVKSTVYGRTNSITCNTNRPFSEVTFT NCALRITTIADRCNGLKECELKTDLLGNPDPCFG TYKYYNTTYDCINGHRVVICEQGYSTLDCGSDSIE IINANYGRGNSRTCSNGILSSQTQNTNCYAPNTL SIVAAMCKGKKTCTVEASNTIFNDPCVGTVKYLT VSYICTREIVTCESSTATLNCGAHRIKIISANYGRT DSTTCSSGRPASQTSNKNCYTPDALNKIAARCE EQSSCEVPATNVVFSDPCFGTYKYLTIVYSCV (GenBank: AHJ14694.1) 53 |
|||
| 1 | 0 | 5 | -5 | 29 | Yes | Yes | MMLFLKPLHLNYFSSWLNGFWREYDYLLQ | |||
| 2 | 0 | 6 | -6 | 306 | No | No | MMLFLSLFTLIISAPGLMVSGENMITCYSDVQRLT CETGLIKVKSTVYGRTNSITCNTNRPFSEVTFTN CALRITTIADRCNGLKECELKTDLLGNPDPCFG TYKYYNTTYDCINGHRVVICEQGYSTLDCGSDSI EIINANYGRGNSRTCSNGILSSQTQNTNCYAPNT LSIVAAMCKGKKTCTVEASNTIFNDPCVGTVKYL TVSYICTREIVTCESSTATLNCGAHRIKIISANYGR TDSTTCSSGRPASQTSNKNCYTPDALNKIAARC EEQSSCEVPATNVVFSDPCFGTYKYLTIVYSCV |
|||
| 3 | 0 | 12 | -12 | 304 | No | No | MILSLFTLIISAPGLMVSGENMITCYSDVQRLTCE TGLIKVKSTVYGRTNSITCNTNRPFSEVTFTNCA LRITTIADRCNGLKECELKTDLLGNPDPCFGTYK YYNTTYDCINGHRVVICEQGYSTLDCGSDSIEIINA NYGRGNSRTCSNGILSSQTQNTNCYAPNTLSIVA AMCKGKKTCTVEASNTIFNDPCVGTVKYLTVSYI CTREIVTCESSTATLNCGAHRIKIISANYGRTDST TCSSGRPASQTSNKNCYTPDALNKIAARCEEQS SCEVPATNVVFSDPCFGTYKYLTIVYSCV |
|||
| 4 | 0 | 16 | -16 | 6 | Yes | Yes | MMLFLN | |||
| 5 | 0 | 183 | -183 | 8 | No | Yes | MMLFLKRI (Assuming splicing is not altered) | |||
| 6 | 1 | 183 | -182 | 5 | Yes | Yes | MMLFL (Assuming splicing is not altered) | |||
| 7 | 1 | 0 | 1 | 31 | Yes | Yes | MMLFLKTQPLHLNYFSSWLNGFWREYDYLLQ | |||
| 8 | 1 | 0 | 1 | 31 | Yes | Yes | MMLFLKSQPLHLNYFSSWLNGFWREYDYLLQ | |||
| 9 | 20 | 0 | 20 | 18 | Yes | Yes | MMLFLKASSHISSSASSP | |||
Table 2: Effects of indel mutations in the rhamnose binding lectin (RBL) gene of channel catfish, Ictalurus punctatus, on predicted protein length (amino acids) and sequence compared to the wild type sequence (WT).
