Beaucoup de processus biologiques se produire sur les sites des interactions cellule-cellule, par exemple, cellules adhérence1,2,3, cellule-cellule fusion4 et reconnaissance cellulaire5. Ces événements sont particulièrement importants lors du développement des organismes pluricellulaires et pour la communication de cellule-cellule, par exemple, au cours de la réponse immunitaire. Ces processus sont généralement véhiculées par des protéines qui sont localisés à la surface, c’est-à-dire à la membrane plasmique (PM) des cellules voisines et subissent des interactions spécifiques au contact cellule-cellule qui sont précisément réglementées dans l’espace et le temps. Dans de nombreux cas, ces interactions sont directement homo – ou des interactions protéine-protéine hétérotypiques trans , mais peuvent aussi comporter des ions ou des ligands agissant comme linkers extracellulaire1. Bien que d’une importance fondamentale, il y a un manque d’essais probing ces interactions protéine-protéine spécifique directement dans l’environnement natif des cellules vivantes. Plusieurs méthodes nécessitent soit désorganisation cellulaire (par exemple, des tests biochimiques comme co-immunoprécipitation6), la fixation (par exemple, certaines des techniques de microscopie optique de Super-résolution et microscopie électronique de cellules contacts7), ou sont non spécifiques, par exemple, agrégation / adhérence dosages8,9. Pour résoudre ce problème, techniques de fluorescence ont été appliquées à l’issu de la fluorescence resonance energy transfert (FRET)10 ou fluorescence complémentation11. Toutefois, pour atteindre suffisamment petites distances entre les fluorophores, ces méthodes nécessitent des étiquettes fluorescentes du côté extracellulaire des protéines10, potentiellement interférer avec des interactions de trans .
Nous présentons ici une analyse alternative de basés sur la fluorescence pour interactions protéine-protéine à contacts cellule-cellule. Cette approche combine des approches de corrélation de fluorescence (balayage spectroscopie de corrélation de fluorescence (sFCCS), nombre de corrélation croisée et la luminosité (ccN & B)) et le mélange des cellules exprimant une construction de fusion de la protéine de intérêt, par exemple, un récepteur d’adhésion. Les récepteurs étudiés dans les deux cellules qui interagissent sont étiquetés avec deux protéines fluorescentes spectralement séparés (FPs), de l’intracellulaire (voir Figure 1 a).
Les méthodes employés reposent sur l’analyse statistique des variations de fluorescence induite par le mouvement diffusif fluorescent des protéines de fusion par le biais du volume focal d’un confocal laser scanning microscope. Plus en détail, le test des sondes la diffusion conjointe des protéines d’intérêt dans les deux voisins PMs à contacts cellule-cellule. Si les protéines subissent des interactions de trans , ces complexes trans portera des protéines fluorescentes émettant dans les deux canaux spectraux, provoquant des fluctuations de corrélation de fluorescence des deux émetteurs. En revanche, si aucune liaison se produit, les fluctuations des protéines numéros face à PMs sera indépendantes, ne causant aucune fluctuation corrélées. L’acquisition peut être effectuée de deux façons : 1) sFCCS repose sur une analyse en forme de ligne au travers le contact de cellule-cellule et sondes efficacement les interactions dans un endroit situé dans la région de contact. Grâce à une analyse temporelle des variations de fluorescence, sFCCS renseigne aussi dynamique, c’est-à-dire, les coefficients de diffusion des complexes protéiques ; 2) ccN & B repose sur une analyse d’un d’une séquence d’images acquises dans les régions de contact cellule-cellule. Il a une capacité à sonder et le long de l’ensemble des interactions de carte contact région (dans un plan focal), mais ne fournit pas d’informations sur la dynamique. Les deux méthodes peuvent être combinées avec une analyse de la luminosité moléculaire, c’est-à-dire le signal de fluorescence moyenne émise dans l’unité de temps par simple diffusion complexes de protéines et, ainsi, fournir des estimations de la stoechiométrie des complexes protéiques au contacts de cellule-cellule.
Dans cet article, nous fournissons des protocoles détaillés pour la préparation des échantillons, d’étalonnage des instruments, d’acquisition de données et d’analyse effectuer l’essai présenté sur un laser confocal commercial microscope à balayage. Les expériences peuvent être effectuées sur n’importe quel instrument équipé de comptage de photons ou détecteurs analogiques et un objectif à grande ouverture numérique. Plus loin, nous discutons des étapes critiques du protocole et fournissent des systèmes de correction pour plusieurs processus causant des fluctuations artéfactuelles signal, par exemple, bruit de détecteur, photoblanchiment ou cellule mouvement. Développé à l’origine pour sonder les interactions entre cellules adhérentes, l’essai peut être modifié pour les cellules en suspension, ou adaptée aux systèmes à membrane modèle, par exemple, les vésicules unilamellaires géant (GUVs) ou le plasma géant vésicules de la membrane (GPMVs), ce qui permet la quantification des interactions dans des environnements différents lipides ou en l’absence d’un cytosquelette organisé12,13.
Spectroscopie de corrélation de fluorescence de balayage est une version modifiée de la spectroscopie de corrélation de fluorescence14 et a été spécialement conçu pour sonder la lente par diffusion dynamique de membranes lipidiques15. Il repose sur une acquisition de balayage ligne perpendiculaire à la PM contenant les protéines fluorescentes d’intérêt. Pour sonder les interactions entre les deux espèces de protéines marquées différemment, l’acquisition est effectuée en deux canaux spectraux en utilisant deux lignes laser et deux fenêtres de détection de fluorophores spectralement séparés. En raison de la dynamique de diffusion lente des protéines dans la MP (D≤ ~ 1 µm2/s), une mesure cross-talk-gratuit peut être effectuée en alternance le régime de l’excitation de la ligne à la ligne15. L’analyse commence par : 1) l’alignement algorithme correction de mouvement latéral cellulaire basé sur block-wise avec une moyenne de ~ 1000 lignes, 2) Détermination de la position avec fluorescence maximum signal, c’est-à-dire la PM position dans chaque bloc et 3) shifting de tous les blocs d’une commune origine12,15, séparément pour chaque voie. Puis, une sélection automatique des pixels correspondant à la PM est exécutée en sélectionnant la zone centrale d’un ajustement gaussien de la somme de toutes les lignes alignées (c.-à-d., Centre ± 2.5σ). Intégration du signal de chaque ligne donne la série temporelle de fluorescence membranaire f (t) dans chaque canal (g = vert canal, r = canal rouge). Notez que la taille du pixel doit être assez petit par exemple, < 200 nm, pour reconstruire la forme du point de fonction d’étalement et de trouver son centre, correspondant à la position de la MP. En présence de photoblanchiment substantiel, les séries chronologiques de fluorescence dans chaque canal peuvent être modélisés par une fonction exponentielle double et puis corrigés avec la formule suivante :16
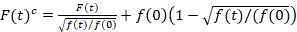 . (1)
. (1)
Il est important de noter que cette formule corrige efficacement tant les amplitudes et les heures de diffusion obtenues par l’analyse de corrélation de f (t)c, par rapport aux estimations de paramètre qui seraient obtenues à partir du non corrigée f (t). Ensuite, les fonctions auto – et la corrélation croisée (ACFs / CCP) de la fluorescence des signaux sont calculées :
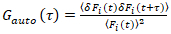 , (2).
, (2).
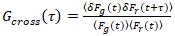 , (3).
, (3).
où δFj’ai = Fi(t) – 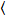 Fi(t)
Fi(t)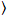 et i = g, r.
et i = g, r.
Un modèle de diffusion bidimensionnelle est ensuite ajusté à toutes les fonctions de corrélation (CFs) :
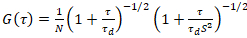 . (4)
. (4)
Ici, N désigne le nombre de protéines fluorescentes dans le volume de l’observation et τd le temps de diffusion pour chaque canal. Ce modèle prend en compte que dans le cadre expérimental décrit, diffusion des protéines dans la MP se produit dans le plan x-z, contrairement à la configuration courante de corrélation de fluorescence spectroscopy (FCS) expériences sur les membranes de palpage diffusion dans le plan x-y du confocal volume17. La taille w0 et le facteur de structure S, décrivant l’allongement wz du volume focal en z, S = wz/w0, sont obtenues à partir d’une mesure de calibration de FCS point effectuée avec colorants spectralement similaires et les mêmes paramètres optiques à l’aide de valeurs déjà disponibles pour le coefficient de diffusion Dcolorant:
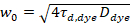 , (5).
, (5).
où τd, colorant est le temps de diffusion moyenne mesurée des molécules de colorant, obtenu à partir d’un modèle pour la diffusion en trois dimensions des données, compte tenu des transitions de compte d’une fraction T de toutes les molécules de N à un état triplet avec une constante de temps ττ:
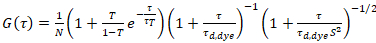 . (6)
. (6)
Enfin, les coefficients de diffusion (D), les valeurs de luminosité moléculaire (ε) et la corrélation croisée relative des données sFCCS (rel.cc.) sont calculées comme suit :
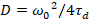 , (7).
, (7).
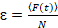 , (8).
, (8).
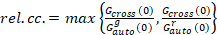 , (9).
, (9).
où GCroix(0) est l’amplitude de la fonction de corrélation croisée et 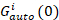 est l’amplitude de la fonction d’autocorrélation dans le canal du je-th.
est l’amplitude de la fonction d’autocorrélation dans le canal du je-th.
Cette définition de la relative corrélation croisée, c’est-à-dire à l’aide de max au lieu de signifier l’équation 9, tient compte du fait que le nombre maximal de complexes des deux espèces de protéines présents à des concentrations différentes est limité par la espèces présentes dans un nombre inférieur.
Luminosité et nombre de corrélation croisée repose sur une analyse de l’instant de l’intensité de fluorescence pour chaque pixel d’une image de pile a acquis au fil du temps à une position fixe dans l’échantillon, généralement composé de ~ 100-200 cadres, avec deux spectrale des canaux () g = vert canal, r = canal rouge). D’après la moyenne temporelle jeet la variance 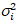 , la luminosité moléculaire εi et numéro nj’ai sont calculées dans chaque pixel et de canaux spectraux (j’ai = g, r)18:
, la luminosité moléculaire εi et numéro nj’ai sont calculées dans chaque pixel et de canaux spectraux (j’ai = g, r)18:
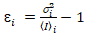 , (10).
, (10).
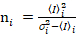 . (11)
. (11)
Il est important de noter que les équations données s’appliquent pour le cas idéal d’un vrai détecteur de comptage de photons. Pour les systèmes de détection analogique, les équations suivantes s’appliquent à19,20:
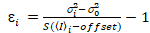 , (12).
, (12).
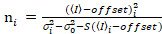 . (13)
. (13)
Ici, S est le facteur de conversion entre les photons détectés et les comtes numériques enregistrées, 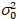 est le bruit de lecture et offset fait référence à l’offset d’intensité de détecteur. Généralement, ces quantités doivent être étalonnées, pour n’importe quel type de détecteur, basée sur la mesure de la variance du détecteur en fonction de l’intensité d’éclairage constant19, par exemple, une surface métallique réfléchissante ou solution de colorant séchées. Le décalage peut être déterminé en mesurant le taux de comptage pour un échantillon sans lumière d’excitation. En effectuant une régression linéaire de la variance associée à détecteur
est le bruit de lecture et offset fait référence à l’offset d’intensité de détecteur. Généralement, ces quantités doivent être étalonnées, pour n’importe quel type de détecteur, basée sur la mesure de la variance du détecteur en fonction de l’intensité d’éclairage constant19, par exemple, une surface métallique réfléchissante ou solution de colorant séchées. Le décalage peut être déterminé en mesurant le taux de comptage pour un échantillon sans lumière d’excitation. En effectuant une régression linéaire de la variance associée à détecteur 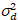 contre le tracé de l’intensité (I), S et peut être déterminée19:
contre le tracé de l’intensité (I), S et peut être déterminée19:
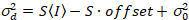 . (14)
. (14)
Enfin, la luminosité de la corrélation croisée est calculée dans chaque pixel et est définie en général comme21
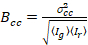 , (15).
, (15).
où 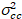 est la Croix et la variance
est la Croix et la variance 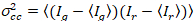 .
.
Pour filtrer les fluctuations à long terme, toutes les ccN & calculs B sont effectuées suivant un wagon couvert, filtrage, indépendamment pour chaque pixel22. En bref, ni, εj’ai (j’ai = g, r) et Bcc sont calculées en glissant des segments de p. ex. cadres de 8-15. Les valeurs ainsi obtenues peuvent être ensuite la moyenne pour obtenir le pixel final les valeurs de nombre et de la luminosité.
Analyse de stoechiométrie
Afin d’estimer la stoechiométrie des complexes protéiques à contacts cellule-cellule, la luminosité moléculaire peut être analysée séparément pour chaque voie spectral pour le sFCCS ou le ccN & B data. Dans sFCCS, une valeur de luminosité est obtenue par la mesure de chaque canal. Dans la ccN & B, on obtient un histogramme de luminosité des pixels situés correspondant au contact cellule-cellule et la valeur moyenne (ou médiane) peut servir de luminosité représentative pour la mesure. En effectuant la même analyse sur une référence monomère, toutes les valeurs de luminosité peuvent être normalisées pour obtenir directement l’État oligomère moyenne des complexes protéine détectée. À ce stade, il est important de corriger la présence de FPs non fluorescent pouvant entraîner une sous-estimation de l’État oligomère. Ceci est généralement effectuée par la mesure de la luminosité d’un homo-dimère référence protéine23,24 utilisant une couleur SCTS-m ou le numéro et la luminosité (N & B).
