Muchos procesos biológicos ocurren en los sitios de las interacciones célula-célula, por ejemplo, célula adhesión1,2,3, célula fusion4 y reconocimiento celular5. Estos eventos son particularmente importantes durante el desarrollo de los organismos multicelulares y para la comunicación de la célula, por ejemplo, durante la respuesta inmune. Estos procesos son típicamente mediados por las proteínas que se localizan en la superficie, es decir, en la membrana plasmática (PM) de los vecinos de las células y sufren interacciones en el contacto de la célula que son precisamente regulado en espacio y tiempo. En muchos casos, estas interacciones son directo homo – o heterotípicos proteínas trans interacciones, pero pueden también implicar iones o ligandos actuando como enlazadores extracelulares1. Aunque de fundamental importancia, hay una falta de análisis de sondeo estas interacciones proteína-proteína específica directamente en el ambiente natural de las células vivas. Muchos métodos requieren o interrupción de la célula (por ejemplo, ensayos bioquímicos tales como co-inmunoprecipitación6), fijación (por ejemplo, algunas de las técnicas de súper-resolución de microscopía óptica y microscopia electrónica de la célula contacto con7), o son no específicos, por ejemplo, la agregación / ensayos de adherencia8,9. Para resolver este problema, se han implementado técnicas de fluorescencia basada en fluorescencia de resonancia energética transferencia (traste)10 o fluorescencia complementación11. Sin embargo, para lograr distancias suficientemente pequeñas entre fluoróforos, estos métodos requieren etiquetas fluorescentes en el lado extracelular de las proteínas10, potencialmente interferir con las interacciones trans .
Aquí, presentamos un ensayo alternativo basado en la fluorescencia para las interacciones proteína-proteína en los contactos célula-célula. Este enfoque combina enfoques de cross-correlación de fluorescencia (análisis espectroscopia de correlación cruzada de fluorescencia (sFCCS), número de la correlación cruzada y brillo (ccN & B)) y la mezcla de células que expresan un concepto de fusión de la proteína de interés, por ejemplo, un receptor de adhesión. Los receptores investigados en las dos células interactuantes están marcados con dos proteínas fluorescentes separadas espectralmente (FPs), desde el intracelular lado (ver figura 1A).
Los métodos empleados se basan en el análisis estadístico de las fluctuaciones de la fluorescencia inducida por el movimiento difusivo de las proteínas fluorescentes de fusión a través del volumen focal de un láser confocal de barrido microscopio. Más detalladamente, el análisis de sondas la difusión conjunta de las proteínas de interés en ambos vecinos PMs en los contactos célula-célula. Si las proteínas se someten a las interacciones trans , estos complejos trans llevará a proteínas fluorescentes emiten en dos canales espectrales, causando fluorescencia correlacionada fluctuaciones de ambos emisores. Por otro lado, si se produce no vinculante, las fluctuaciones del número de proteínas frente a PMs será independientes, no causando fluctuaciones correlacionadas. La adquisición puede realizarse de dos maneras: 1) sFCCS se basa en un análisis en forma de línea a través del contacto célula-célula y efectiva puntas de prueba de las interacciones en un lugar situado en la región de contacto. A través de un análisis temporal de las fluctuaciones de la fluorescencia, sFCCS proporciona también información dinámica, es decir, los coeficientes de difusión de complejos de proteína; 2) ccN & B se basa en un análisis pixel-wise de una secuencia de imágenes adquiridas en las regiones de contacto célula-célula. Tiene capacidad para sondear y mapa interacciones a lo largo de toda la región (en un plano focal) de contacto, pero no proporciona información sobre la dinámica. Ambos métodos pueden combinarse con un análisis de la luminosidad molecular, es decir, la señal de fluorescencia promedio emitida en la unidad de tiempo por solo difunde complejos de proteína y, así, proporcionar estimaciones de la estequiometría de los complejos de proteína en contactos de la célula.
En este artículo, le ofrecemos protocolos detallados para la preparación de la muestra, calibración del instrumento, adquisición de datos y análisis realizar el ensayo presentado en un comercial láser confocal de barrido microscopio. Los experimentos pueden realizarse en cualquier instrumento equipado con conteo de fotones o detectores analógicos y un objetivo con alta apertura numérica. Además discutir pasos críticos del protocolo y proporcionar esquemas de corrección para varios procesos causando fluctuaciones de señal artefactual, p. ej., detector ruido, movimiento fotoblanqueo o celular. Originalmente desarrollado para probar las interacciones entre las células adherentes, el ensayo puede ser modificado para que las células de suspensión, o adaptados a los sistemas modelo de membranas, por ejemplo, propiedades gigantes vesículas (2.fino) o plasma gigante vesículas de membrana (GPMVs), que permite la cuantificación de las interacciones en entornos diferentes lípidos o en la ausencia de un citoesqueleto organizado12,13.
Análisis de espectroscopia de fluorescencia de la correlación cruzada es una versión modificada de la espectroscopia de correlación cruzada de fluorescencia14 y fue diseñado específicamente para sonda lenta dinámica difusión en membranas de lípidos15. Se basa en una adquisición de exploración de línea perpendicular a la PM que contiene las proteínas fluorescentes de interés. Para probar las interacciones de dos especies de proteínas diferentemente etiquetadas, la adquisición se realiza en dos canales espectrales usando dos líneas láser y dos ventanas de detección de fluoróforos espectralmente separados. Debido a la dinámica de la difusión lenta de las proteínas en el PM (D≤ ~ 1 μm2/s), puede realizarse una medición libre de charla Cruz alternando el esquema de excitación de la línea a la línea15. El análisis comienza con: 1) un algoritmo de alineamiento corregir para el movimiento lateral de la célula basado en block-wise con un promedio de ~ 1000 líneas, 2) determinación de la posición con fluorescencia máxima señales decir, el PM posición, en cada bloque y cambio de 3) de todos los bloques a un común origen12,15, por separado en cada canal. A continuación, una selección automática de píxeles correspondiente a la PM se realiza seleccionando la región central de un ajuste gaussiano de la suma de todas las líneas alineadas (es decir, centro ± 2.5σ). Integración de la señal en cada línea produce la membrana fluorescencia serie f (t) en cada canal (g = green canal, r = canal rojo). Nota que el tamaño del pixel ha de ser pequeño, por ejemplo, < 200 nm, para reconstruir la forma del punto de difundir la función y encontrar su centro, correspondiente a la posición de la PM. En presencia de fotoblanqueo substancial, la serie de tiempo de fluorescencia en cada canal puede modelada con una función doble exponencial y luego corregir con la fórmula siguiente:16
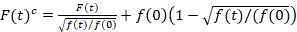 . (1)
. (1)
Es importante tener en cuenta que esta fórmula corrige eficazmente las amplitudes y tiempos de difusión obtenidos del análisis de correlación de f (t)c, en comparación con las estimaciones del parámetro que se obtendría de la no corregida f (t). Entonces, las funciones de auto – y la correlación cruzada (ACFs / CCFs) de la fluorescencia señales se calculan:
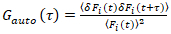 , (2).
, (2).
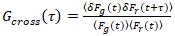 , (3).
, (3).
donde δF = F(t) – 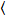 F(t)
F(t)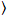 y = g, r.
y = g, r.
Un modelo de difusión bidimensional entonces se acopla a todas las funciones de correlación (SFC):
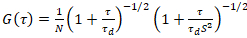 . (4)
. (4)
Aquí N denota el número de proteínas fluorescentes en el volumen de observación y τd el tiempo de difusión para cada canal. Este modelo toma en cuenta que en la configuración experimental descrita, difusión de proteínas en la PM se presenta en el plano x-z, en contraste con la configuración de uso general de correlación de fluorescencia espectroscopia (FCS) experimentos en las membranas de sondeo difusión en el plano x-y del confocal volumen17. La cintura w0 y el factor de estructura S, describiendo la elongación wz del volumen focal en z, S = wz/w0, se obtienen de una medida de calibración punto de FCS realizada con tintes espectral similares y misma configuración óptica utilizando valores ya están disponibles para el coeficiente de difusión Dtinte:
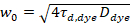 , (5).
, (5).
donde τd, tinte es el tiempo medido medio difusión de las moléculas de colorante, de un modelo de difusión tridimensional de los datos, teniendo en las transiciones de la cuenta de una fracción T de todas las moléculas de N a una Estado triplete con una constante de tiempo ττ:
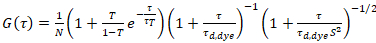 . (6)
. (6)
Por último, coeficientes de difusión (D), los valores de brillo molecular (ε) y la relativa correlación cruzada de datos sFCCS (rel.cc.) se calculan como sigue:
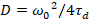 , (7).
, (7).
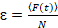 , (8).
, (8).
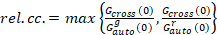 , (9).
, (9).
donde GCruz(0) es la amplitud de la función de correlación cruzada y 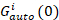 es la amplitud de la función de autocorrelación en el -ésimo canal.
es la amplitud de la función de autocorrelación en el -ésimo canal.
Esta definición de la relativa correlación cruzada, es decir, utilizando máximo en lugar de decir en la ecuación 9, toma en cuenta que el número máximo de conjuntos de dos especies de proteínas presentes en diferentes concentraciones se ve limitado por la especies presentes en un número inferior.
Brillo y el número de la correlación cruzada se basa en un análisis del momento de la intensidad de fluorescencia para cada píxel de una pila de imagen adquirida con el tiempo en una posición fija en la muestra, por lo general consisten en ~ 100-200 marcos, con dos espectral canales () g = green canal, r = canal rojo). De la media temporal Ii y varianza 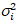 , el brillo molecular εi y número n se calculan en cada pixel y canal espectral ( = g, r)18:
, el brillo molecular εi y número n se calculan en cada pixel y canal espectral ( = g, r)18:
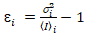 , (10).
, (10).
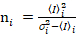 . (11)
. (11)
Es importante tener en cuenta que las ecuaciones dadas se aplican al caso ideal de un verdadero detector de recuento de fotones. Las ecuaciones siguientes aplican para sistemas de detección analógico,19,20:
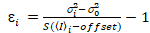 , (12).
, (12).
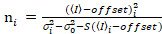 . (13)
. (13)
Aquí, S es el factor de conversión entre fotones detectados y las cuentas de digitales grabadas, 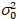 es el ruido de lectura y offset se refiere a la compensación de la intensidad de detector. Por lo general, estas cantidades se deberán calibrar, para cualquier tipo de detector, basado en la medición de la variación del detector en función de la intensidad de iluminación constante19, p. ej., una superficie metálica reflectora o solución colorante seco. El offset puede determinarse midiendo la tasa de conteo para una muestra sin luz de excitación. Realizando una regresión lineal de la varianza asociados detector
es el ruido de lectura y offset se refiere a la compensación de la intensidad de detector. Por lo general, estas cantidades se deberán calibrar, para cualquier tipo de detector, basado en la medición de la variación del detector en función de la intensidad de iluminación constante19, p. ej., una superficie metálica reflectora o solución colorante seco. El offset puede determinarse midiendo la tasa de conteo para una muestra sin luz de excitación. Realizando una regresión lineal de la varianza asociados detector 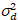 contra la trama de la intensidad (), S y puede ser resuelto19:
contra la trama de la intensidad (), S y puede ser resuelto19:
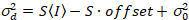 . (14)
. (14)
Por último, el brillo de la correlación cruzada se calcula en cada pixel y se define en general como21
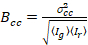 , (15).
, (15).
donde 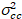 es la varianza de la Cruz
es la varianza de la Cruz 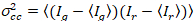 .
.
Para filtrar las fluctuaciones de larga duración, todos ccN y cálculos de B se realizan siguiendo un furgón filtrado, independientemente para cada pixel22. En Resumen, ni, ε ( = g, r) y Bcc se calculan en desplazamiento de segmentos de p. ej., 8-15 marcos. Los valores así obtenidos pueden ser promediados entonces para obtener el pixel final valores de número y brillo.
Análisis de la estequiometría
Para estimar la estequiometría de los complejos de la proteína en los contactos célula-célula, el brillo molecular puede analizarse por separado en cada canal espectral para la sFCCS o ccN y B los datos. En sFCCS, un valor de brillo se obtiene por medición de cada canal. En ccN y B, se obtiene un histograma de brillo de los píxeles correspondientes al contacto célula-célula y el valor promedio (o mediano) puede utilizarse como brillo representativo para la medición. Por realizar el mismo análisis en referencia monomérica, todos los valores de brillo pueden ser normalizados para obtener directamente el estado oligomérico promedio de los complejos de proteína detectado. En este punto, es importante corregir la presencia de FPs no fluorescente que puede resultar en una subestimación del estado oligomérico. Normalmente esto se realiza midiendo el brillo de un homo dimérico referencia proteína23,24 con sFCS un color o número y el brillo (N & B).
