Figure 3 illustrates the results of an experiment with a BW reporter system. In this experiment, CLL cells were pre-incubated with different anti-CD20 antibodies (rituximab and obinutuzumab) and then co-incubated with transfected BW cells which express CD16a-CD3ζ. Similar experiments were conducted in our study9. Figure 3A presents an image of a raw ELISA plate where the color intensity corresponds to a concentration of mIL-2. The experiment was performed in triplicate. This experiment included several controls: CLL cells incubated with parental untransfected BW cells (upper row) and CLL cells incubated with BW cells which express CD16a-CD3ζ but without an antibody or with a control antibody (six wells in the second row on the left). The incubation of BW cells that expressed CD16 with CLL cells that were pre-incubated with anti-CD20 antibodies induced a significant secretion of mIL-2 compared to the mIL-2 level of the control wells. Importantly, pre-incubation of the CLL cells with the anti-CD20 obinutuzumab activated CD16 more strongly than rituximab, a known finding which was recapitulated by our system. The last row shows descending pre-defined concentrations of recombinant mIL-2 (without the addition of cells). The right well in the last row does not have mIL-2 and represents the background reading, which appears similar to the control wells. Figure 3B presents the quantification of the results of the ELISA plate shown in Figure 3A. The optical density (OD) levels were converted into mIL-2 concentrations based on the standard curve with recombinant mIL-2. Figure 3C presents a dose response experiment where different doses of antibodies were pre-incubated with CLL cells and then incubated with BW cells expressing CD16.
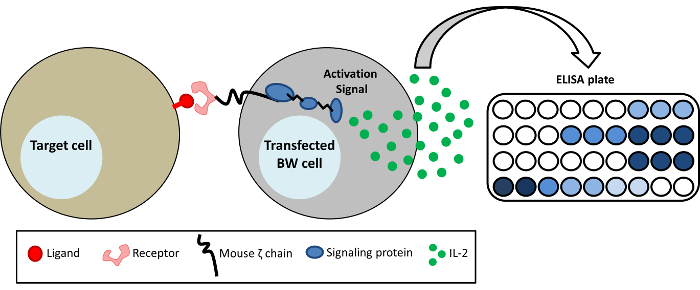
Figure 1: Schematic representation of the BW reporter system. BW5147 cells are stably transfected with the extracellular portion of different receptors fused to the transmembrane and cytoplasmic domains of the mouse CD3ζ chain (chimeric recptor-CD3ζ). Activation of a specific receptor-CD3ζ by a ligand results in a secretion of mIL-2 which can be detected by ELISA. Please click here to view a larger version of this figure.
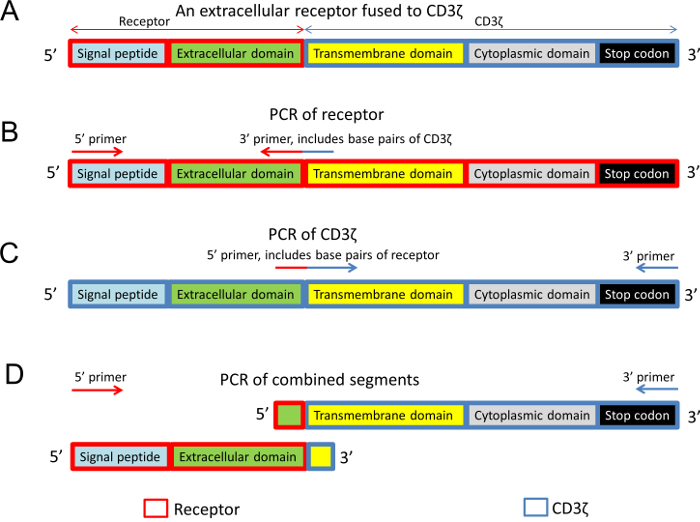
Figure 2: The structure of the chimeric receptor-CD3ζ and the design of PCR primers. (A) General structure of an extracellular receptor fused to the transmembrane and intracellular domains of mouse CD3ζ. (B) The extracellular domain of the receptor was amplified with a 3' primer which also included nucleotides of CD3ζ. (C) The intracellular and transmembrane domains of CD3ζ were amplified with a 5' primer that also included receptor nucleotides. (D) In the final PCR reaction, both segments, which have overlapping sequences, were used as the DNA template. In all figure panels, different protein domains are color-coded and boxed in blue rectangles (CD3ζ sequence) and red rectangles (the receptor sequence). Please click here to view a larger version of this figure.
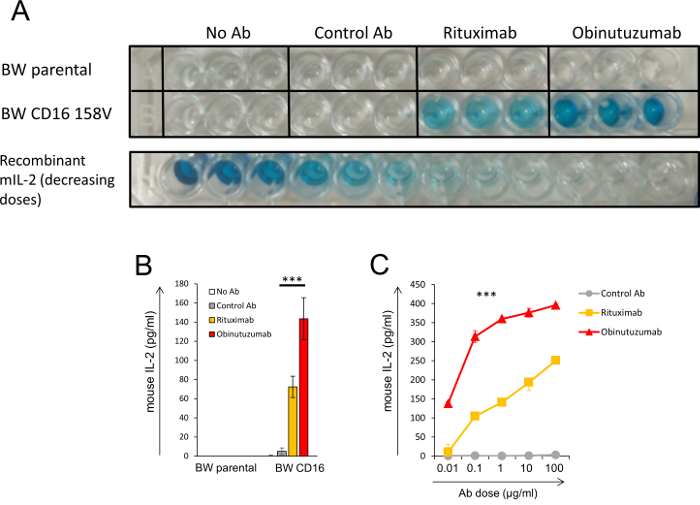
Figure 3: Illustration of the final product of the BW reporter system. (A, B, C) CLL cells were pre-incubated with or without two anti-CD20 antibodies (rituximab and obinutuzumab) or a control antibody. Afterwards, the antibody-bound cells were incubated with parental BW cells or BW cells which express CD16a-CD3ζ. The level of mIL-2 in the supernatant was determined by ELISA. (A) An image of a raw ELISA plate. Color intensity indicates the concentration of mIL-2 in the supernatant. The experiment was performed in triplicate. The lower row shows the ELISA readout in decreasing concentrations of recombinant mIL-2 which were analyzed in the same plate. (B) Quantification of the ELISA reading shown in (A). The mIL-2 levels were calculated using a standard curve of mil-2. (C) A dose response experiment where different doses of antibodies were pre-incubated with CLL cells and then incubated with BW cells expressing CD16a-CD3ζ. ***, P < 0.001; Student's t test (B) or ANOVA with multiple comparisons (C). The only comparison in (C) which was not significantly different was for rituximab and the control Ab in the first concentration (0.01 µg/mL). Error bars represent the standard deviation of the triplicates. Similar experiments were conducted as part of one of our recent study9. Please click here to view a larger version of this figure.
