NOTE: The following steps were performed using IIA1.6 B cells.
1. B cell activation with antigen-coated beads
- Preparation of antigen-coated beads
- To activate B cells, use NH2-beads covalently coated with antigen (Ag-coated beads), which are prepared using 50 µL (~20 x 106 beads) of 3 µm NH2-beads with activating (BCR-ligand+) or non-activating (BCR-ligand-) antigens.
- For IIA1.6 B cells use anti-IgG-F(ab')2 fragment as BCR-ligand+ and anti-IgM-F(ab')2 or bovine serum albumin (BSA) as BCR-ligand-. To activate primary B cells or IgM+ B cell line use anti-IgM-F(ab')2 as BCR-ligand+ and BSA as BCR-ligand-.
NOTE: To avoid the binding of ligands to the Fc receptor, use F(ab') or F(ab')2 antibodies fragments instead of full-length antibodies. - To proceed with the preparation of Ag-coated beads, place the beads in low protein binding microcentrifuge tubes to maximize the beads recovery during the sample manipulation. Add 1 mL of 1x phosphate-buffered saline (PBS) to wash beads and centrifuge at 16,000 x g for 5 min. Aspirate the supernatant.
- Resuspend the beads with 500 µL of 8% glutaraldehyde to activate the NH2 groups and rotate for 4 h at room temperature (RT).
CAUTION: The glutaraldehyde stock solution should only be used in a chemical fume hood. Follow the instructions on the material safety data sheet (MSDS). - Centrifuge beads at 16,000 x g for 5 min, remove the glutaraldehyde and wash the beads three more times with 1 mL of 1x PBS.
CAUTION: The glutaraldehyde solution should be discarded as a hazardous chemical waste. - Resuspend the activated beads in 100 µL of 1x PBS. The sample can be divided into two low protein binding microcentrifuge tubes: 50 µL for the BCR-ligand+ and 50 µL for BCR-ligand-.
- To prepare the antigen solution use two 2 mL low protein binding microcentrifuge tubes containing 100 µg/mL of antigen solution in 150 µL of PBS: One tube with BCR-ligand+ and other with BCR-ligand-.
- Add 50 µL of activated beads solution to each tube containing 150 µL of antigen solution, vortex and rotate overnight at 4 °C.
- Add 500 µL of 10 mg/mL BSA to block the remaining reactive NH2 groups on the beads and rotate for 1 h at 4 °C.
- Centrifuge the beads at 16,000 x g for 5 min at 4 °C and remove the supernatant. Wash the beads with cold 1x PBS three more times.
- Resuspend the activated beads in 70 µL of 1x PBS.
- To determine the final concentration of Ag-coated beads, dilute a small volume of beads in PBS (1:200) and count using a hemocytometer. Then store at 4 °C until use.
NOTE: Ag-coated beads should not be stored for more than 1 month.
- Preparation of poly-L-lysine coverslips
- Before the B cell activation assay with Ag-coated beads, prepare poly-L-lysine-coated coverslips (PLL-coverslips). Use a 50 mL tube containing 40 mL of 0.01% w/v of PLL solution and immerse the 12 mm-diameter coverslips into the solution. Rotate overnight at RT.
- Wash the coverslips with 1x PBS and leave to dry on a 24-well plate lid covered with paraffin film. Proceed with B cell activation.
- B cell activation
- To start B cell activation with beads, first dilute the IIA1.6 B cell line to 1.5 x 106 cells/mL in CLICK medium (RPMI-1640 supplemented with 2 mM L-Alanine-L-Glutamine + 55 µM beta-mercaptoethanol + 1 mM pyruvate + 100 U/mL Penicillin + 100 µg/mL streptomycin) + 5% heat-inactivated fetal bovine serum [FBS]).
- Combine 100 µL (150,000 cells) of IIA1.6 B cells with Ag-coated beads at 1:1 ratio in 0.6 mL tubes. Mix gently using a vortex and seed onto PLL-coverslips. Incubate for different time points in a cell culture incubator (37 °C / 5% CO2). The typical activating time points are 0, 30, 60 and 120 min. For time 0, place the PLL-coverslips into the 24-well plate lid on ice. Add the cells-Ag-coated bead mixture and incubate for 5 min on ice.
NOTE: It is important to mix by vortex instead of pipetting up and down, because this could reduce the number of beads in the sample, due to their accumulation in the plastic tip of the pipette. Use beads coated with BCR-ligand- as a negative control for each assay. We recommend activating the longest incubation time first and then the following time points. Calculate intervals of activation for samples such that they are ready for fixation at the same time. - Add 100 µL of cold 1x PBS to each coverslip to stop the activation and continue with the immunofluorescence protocol. See protocol section 3.
2. B cell activation on antigen-coated coverslips
- Preparation of antigen-coated coverslips
- Before activation, prepare the antigen solution (1x PBS containing 10 µg/mL BCR-ligand+ and 0.5 µg/mL rat anti-mouse CD45R/B220).
NOTE: Consider preparing the coverslips the day before the assay. B220 improves B cell adhesion, however, the BCR-Ligand+ is sufficient to generate the spreading response. - Place the 12 mm coverslip onto a 24-well plate lid covered with paraffin film, add 40 µL of antigen solution onto each coverslip and incubate at 4 °C overnight. Seal the plate to avoid evaporation of antigen solution.
- Wash the coverslips with 1x PBS and air dry.
- Before activation, prepare the antigen solution (1x PBS containing 10 µg/mL BCR-ligand+ and 0.5 µg/mL rat anti-mouse CD45R/B220).
- B cell activation
- To start the activation, dilute IIA1.6 B cells to 1.5 x 106 cells/mL in CLICK medium + 5% FBS.
- Add 100 µL of cells onto an antigen-coated coverslip (Ag-coverslip) and activate for different time points in a cell incubator at 37 °C / 5% CO2. The typical activating time points are 0, 30 and 60 min.
NOTE: As in section 1, we recommend starting with the longest activating time point in order to fix all the samples at the same time. - For time 0, place the 24-well plate lid containing the Ag-coverslip on ice and add the cells. Incubate for 5 min on ice.
- Carefully aspirate the media on each Ag-coverslip and then add 100 µL of cold 1x PBS to stop the activation. Continue with the immunofluorescence protocol. See section 3.
3. Immunofluorescence
NOTE: To avoid cross-reactivity of the secondary antibody with the BCR of IIA 1.6 B cells, do not use mouse-derived primary antibodies.
- Remove the 1x PBS and proceed with the fixation of each coverslip.
NOTE: To decide which fixation medium to use, check the antibody/dye data sheet. For example, for actin labeling by phalloidin use paraformaldehyde (PFA) fixation medium, and for the centrosome labeling by pericentrin use methanol fixation.- Add 50 µL of 1x PBS supplemented with 4% PFA and incubate for 10 min at RT.
CAUTION: Formaldehyde is toxic. Please read the MSDS before working with this chemical. PFA solutions should be only made under a chemical fume hood wearing gloves and safety glasses. PFA solution should be discarded as a hazardous chemical waste. - Alternatively, add 50 µL of cold methanol and incubate for 20 min.
- Add 50 µL of 1x PBS supplemented with 4% PFA and incubate for 10 min at RT.
- Wash three times with 1x PBS.
NOTE: Coverslips can be stored at 4 °C at this point for maximum three days in 1x PBS. - Remove 1x PBS and add 50 µL of blocking buffer (2% BSA + 0.3 M glycine in 1x PBS) onto each coverslip. Incubate at RT for 10 min.
- Aspirate gently and add 50 µL of permeabilization buffer (PB) (0.2% BSA + 0.05% saponin in 1x PBS). Incubate at RT for 20 min.
- Prepare the antibodies or dyes in PB (use 30 µL per coverslip) and incubate at RT for 1 h or overnight at 4 °C. Refer to the Table of Materials for additional details.
- To label the microtubule organizing center (MTOC) or centrosome use the following antibodies: anti-γ-Tubulin (1:500), anti-Cep55 (1:500), anti-α-tubulin (1:500), anti-pericentrin (1:1000).
NOTE: For centrosome labeling B cells can be transfected with a centrin-GFP expression plasmid. - For labeling Golgi apparatus use anti-Rab6a (1:500).
NOTE: Other antibodies or dyes can also be used. - For labeling lysosomes, use anti-Lamp1 (1:200).
NOTE: Anti-H2-DM and anti-MHC-II also can be used to label antigen processing compartments15,16. - For labeling endoplasmic reticulum use anti-Sec61a (1:500).
- For actin cytoskeleton consider the following: polymerized actin can be visualized by Phalloidin conjugated to fluorescent dyes.
NOTE: B cells transfected with a LifeAct-GFP/RFP expression plasmid can also be used to label actin.
- To label the microtubule organizing center (MTOC) or centrosome use the following antibodies: anti-γ-Tubulin (1:500), anti-Cep55 (1:500), anti-α-tubulin (1:500), anti-pericentrin (1:1000).
- Wash the coverslips three times with PBS.
- Dilute the secondary antibody or dyes in PBS (use 30 µL per coverslip) and incubate 1 h at RT.
NOTE: Avoid exposing the samples to direct light to preserve the quality of the fluorescence signal. - Wash the coverslips three times with PBS.
- Remove the PBS solution from coverslips.
- Add 4 µL of mounting reagent to a microscope slide. Mount the coverslip onto the slide with the cell side facing down. Allow the slides to dry for 30 min at 37 °C or at RT overnight.
NOTE: Consider using an "anti-fade" mounting reagent (see the Table of Materials). - Acquire fluorescence images on a confocal or epifluorescence microscope with a 60x or 100x oil immersion objective. For each acquisition consider the transmitted light or bright field to easily identify B cells interacting with beads.
NOTE: Take three-dimensional (3D) images, covering the entire cell using z-stacks. We recommend taking 0.5 µm thick stacks, however this depends on the microscope.
4. Image analysis
NOTE: The following algorithms are described for ImageJ software. However, this can be performed using an equivalent software. Also, consider that for all fluorescence intensity measurements we use the integrated fluorescence density ("RawIntDen" in ImageJ), because this parameter considers the total amount of fluorescence in each pixel of the image, taking the area into account.
- Analysis of the distribution of organelles in B cells activated with Ag-coated beads
NOTE: To quantify the polarization of cell components to the IS, we define an arbitrary value as a measure of proximity to the IS. The index ranges between -1 (anti-polarized) and 1 (Fully polarized, object on the bead), as was previously presented by Reversat et al.12.- Estimate the polarity index for the centrosome and Golgi apparatus (Figure 1B).
NOTE: This algorithm can be used for organelles that are confined to a one point.- First define the bead and cell areas to analyze using the circle tool selection to delimit the boundaries of both, and then save them as regions of interest (ROI). See Figure 1, Figure 2, Figure 3, and Figure 4.
- Determine the cell center (CC) and the bead center (BC) by running Analyze | Measure on cell and bead areas respectively. X and Y values obtained from the Résultats window determine the center coordinates.
- Manually determine the center of the centrosome or Golgi apparatus (Organelle) using the point tool selection in ImageJ and run Analyze | Measure. X and Y values obtained from the Résultats window determine the coordinates.
- Then, draw an angle from CC to Organelle (a) and CC to BC (b) using the angle tool selection and run Analyze | Measure. The angle value in the results window shows the angle (α) between both vectors (a and b).
- Calculate the polarity index using the following formula:
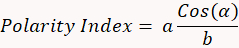
- Estimate the polarity index for lysosomes (Figure 1F).
NOTE: We use this algorithm to analyze the polarity of organelles that display a more dispersed distribution, such as lysosomes.- Define the bead and cell areas to analyze, using the circle tool selection to delimit the boundaries of both, and save them as ROI. Once the bead and cell areas have been determind, set the fluorescence channel and project the image into one z-stack (Image | Stacks | Z-Project [Sum slices]), then run Analyze | Measure and extract the mass center (MC) coordinates (MX and MY) from the results windows.
- Apply the same algorithm mentioned before changing Organelle for MC. Thus, the angle (α) is defined by CC-MC (a) and CC-BC (b).
- Calculate the polarity using the following formula:
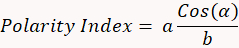
- Estimate lysosome recruitment to the synaptic area (Figure 2B).
NOTE: This algorithm is used to quantify an organelle adjacent to the IS.- Once the bead and cell areas have been determined, determine the angle of the vector between the CC and BC and then rotate the image to achieve a 0° angle.
- Set the fluorescent channel and project the image into one z-stack (Image | Stacks | Z-Project [Sum slices]), eliminate all the fluorescence that falls outside the cell and bead area together (run edit | clear outside in ImageJ).
- Using the rectangle tool selection, draw a rectangle adjacent to the bead and measure the synaptic area fluorescence (SAF). This rectangle is a quarter of the cell width.
- Select the entire image and run Analyze | Measure to obtain the whole cell fluorescence (WCF).
- Calculate the organelle fluorescence percentage adjacent to the IS using the following formula:

- Quantify recruitment of cellular components to the centrosome.
NOTE: This algorithm, adapted from Obino et al.5, is used to quantify the enrichment of organelles at the centrosome area. Briefly, we consider the centrosome area as the domain in which the centrosome-associated organelle fluorescence remains constant or above 70% in the fluorescence/radius plot. It is essential to set this parameter at resting conditions because this radius could change upon activation.- Once bead and cell areas have been determined, define the localization of the centrosome using the point tool selection (Figure 3B).
- First, determine the maximum area around the centrosome that is possible to quantify, by drawing a 3 µm-radius circle surrounding the centrosome.
- Use the ImageJ plugin Radial Profile, which measures the fluorescence in concentric circles and displays a fluorescence/radius plot.
- Identify the maximum radius within which at least 70% of the fluorescence intensity is maintained.
- Calculate the fluorescence density ratio using the following formula:
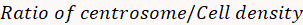 =
= 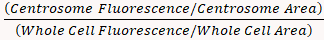
NOTE: The fluorescence density ratio indicates the concentration of an organelle at the centrosome compared to its distribution in the whole cell.
- Estimate the polarity index for the centrosome and Golgi apparatus (Figure 1B).
- Analysis of cell spreading and distribution of organelles in B cells activated on Ag-coverslips
- Estimate organelle distribution at the IS (Figure 4B).
NOTE: This algorithm allows the quantification of the XY distribution of organelles and their concentration at the center of the IS. We define a ratio of fluorescence density between the central and the total synaptic area. The values obtained can vary from negative to positive, indicating a peripheral or a central distribution of the organelle, respectively.- Determine the slice in which the cell is in contact with the cover.
NOTE: To delimit the cell boundaries one can label the plasma membrane or actin which is enriched in the periphery of the IS. - Change the type of the slice to 8-bit, then binarize (Process | Binary | Make Binary) and connect the nearest outsider points Process | Binary | Outline. Delimit the boundaries of the cell area (CA) using the polygon tool selection.
NOTE: This step is useful to increase the contrast of the cell boundaries and make it easier to identify. Also, at this point, it is possible to apply the Analyze particle plugin of ImageJ to determine the CA automatically. However, other cells in the same field can interfere with the results. - Take CA parameters (height and width) and delimit a central rounded area, which is separated from the boundaries by a quarter of the height and width values, consider this area as the Cell Center Area ( CCA).
- Calculate the fluorescence density distribution at the center of the IS using the following formula:

- Determine the slice in which the cell is in contact with the cover.
- Measure organelle distribution in Z planes (Figure 4C).
NOTE: This analysis determines the general distribution of organelle fluorescence across Z planes of B cells activated onto Ag-coverslips, showing the percentage of fluorescence per Z fraction.- To quantify the fluorescence distribution in Z, first determine the plane of the IS where the cell is in contact with the Ag-coverslip and then the plane corresponding to the upper limit of the cell.
- Draw a line across the cell center.
- Reslice the image in Z (Image | Stacks | Reslice), to obtain an XZ image.
- Measure the height and divide the XZ image into 10 consecutive rectangles of the same height (Z fraction) from the bottom (IS interface) to the top (upper side of the cell) and quantify the fluorescence signal in each one.
- Normalize the fluorescence intensity of each Z fraction by the sum of the total fluorescence of the 10 fractions.
- Plot the percentage of fluorescence intensity per Z fraction of the cell.
- Estimate organelle distribution at the IS (Figure 4B).
5. Isolation of centrosome-enriched fraction from resting and activated B cells
NOTE: Keep all solutions at 4 °C during the experiment to avoid protein degradation. This protocol was adapted from previous work17,18.
- Activate 2 x 107 B cells with Ag-coated beads in 2 mL of CLICK medium + 2% heat-inactivated FBS (ratio 1:1). Consider non-activated B cells as resting B cells.
- Add cytochalasin D (2 µM) and nocodazole (0.2 µM) and incubate for 1 h at 37 °C.
NOTE: These drugs are used to gently detach the centrosome from the nucleus, by depolymerizing actin cytoskeleton and microtubules, respectively, to avoid nuclear contamination. - Wash each sample with 5 mL of cold 1x TBS (50 mM Tris-HCl, pH 7.6, 150 mM NaCl) and then with 1 mL of 0.1x TBS supplemented with 8% sucrose.
- Resuspend the cells with 150 µL of centrosome lysis buffer (1 mM HEPES, pH 7.2, 0.5% NP-40, 0.5 mM MgCl2, 0.1% beta-Mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride (PMSF) or protease inhibitor cocktail) and pipette up and down until viscosity decreases, which indicates cell lysis is identified in the sample. Put the sample in a 1.5 mL tube.
- Centrifuge at 10,000 x g for 10 min at 4 °C, to separate the organelles from the nucleus.
- Carefully recover the supernatant and place it on top of a 1.5 mL tube with 300 µL of gradient buffer (GB) (10 mM PIPES pH 7.2, 0.1% Triton X-100, 0.1% beta-mercaptoethanol) containing 60% sucrose.
- Centrifuge at 10,000 x g for 30 min at 4 °C to concentrate the centrosomes in the 60% sucrose fraction.
- Meanwhile, prepare a discontinuous gradient in 2 mL ultracentrifuge tubes by overlaying 450 µL of GB + 70% sucrose with 270 µL of GB + 50% sucrose and then 270 µL of GB + 40% sucrose.
- After the first centrifugation (centrosome-concentrated fraction), discard the upper fraction (less dense portion) until reaching the interface and vortex the remaining sample in the tube. Then, overlay on top of the discontinuous gradient previously prepared with the centrosome enriched sample.
NOTE: Be careful not to disrupt the gradient, all pipetting procedures must be carefully performed. - Centrifuge at 40,000 x g for 1 h at 4 °C with minimal acceleration and with the centrifuge brake set to off, to avoid disrupting the gradient.
- Collect 12 fractions of 100 µL into separate tubes beginning from the top.
- Identify the centrosome enriched fractions by immunoblot using γ-tubulin as the centrosome marker.
NOTE: We usually find centrosome-enriched extracts between fractions 6 and 8.
The present article shows how B cells can be activated using immobilized antigen on beads or coverslips to induce the formation of an IS. We provide information on how to identify and quantify the polarization of different organelles by immunofluorescence and how to characterize proteins that undergo dynamic changes in their association to the centrosome, which polarizes to the IS, using a biochemical approach.
The imaging of B cells by immunofluorescence allows us to follow the dynamics of organelles such as the centrosome, Golgi apparatus and lysosomes, which are recruited to the IS upon B cell activation. One can obtain quantitative parameters to measure the polarization of these organelles to the IS and compare them under different conditions. As we show in Figure 1A, the Centrosome and Golgi Apparatus are recruited to the IS upon B cell activation. Recruitment was not observed in B cells stimulated with BCR-ligand- indicating that BCR engagement is required to mobilize the centrosome and Golgi apparatus to the IS (Figure 1A). To obtain a polarity index for each organelle, as a measurement of its proximity to the IS, we considered three features: the distance between the organelle and the center of the cell, the distance between the bead center and the cell center, and the angle between these two vectors (Figure 1B). This index ranges between -1 (anti-polarized) and 1 (fully polarized, object on the bead). Figure 1C and 1D show graphs where polarity indexes of each organelle are plotted versus time of activation. In agreement with immunofluorescence staining, both the centrosome and Golgi apparatus, display more positive polarity indexes upon longer time points of activation, reflecting how they are progressively recruited to the IS. This algorithm is applicable for organelles confined to one point.
Additionally, we quantified the polarization of organelles that display a dispersed distribution, such as lysosomes (Figure 1E), by applying the same algorithm mentioned before, but changing the point coordinate by the mass center coordinates of lysosomes (Figure 1F). We can observe that the polarity indexes of lysosome pools reach more positive values upon activation, which indicates that the lysosomes are being mobilized towards the synapse upon B cell activation (Figure 1G).
We also defined two algorithms to determine organelles that are in contact with the synaptic interface (bead) and the amount of organelles at the proximity of the synapse, but not necessarily in contact. As we show in Figure 2, we quantified lysosomes contacting the synaptic interface (Figure 2A), by dividing the LAMP-1 signal at the bead area by the total fluorescence in the cell plus the bead (Figure 2B). It is important to consider that the diameter of the bead area determines the ranges of the percentages obtained. For example, in the quantification displayed (Figure 2B), we draw a circle with a 3 µm diameter to measure the fluorescence at the bead, which is a restrictive parameter considering the bead size (3 µm). Using this parameter, the results show that lysosomes are progressively recruited to the synaptic interface upon B cell activation, reaching 10-15% of the total mass (Figure 2C). Another approach shown is the quantification of lysosomes to the synaptic area. Figure 2D shows that lysosomes progressively accumulate up to 25% of their lysosomes at the IS. Overall, by performing both types of analysis one can evaluate the polarization and docking of organelles at the IS.
During B cell activation, changes in the pool of centrosome-associated proteins have been documented5. For instance, one of these proteins that changes their association at the Centrosome during B cell activation is actin. In this case actin becomes depleted within this region, which allows the centrosome to become detached from the nucleus, promoting its polarization to the IS5. Here we present the quantification of actin at the Centrosome and how it is depleted upon B cell activation (Figure 3). To define the centrosome area used to quantify associated actin, we measured the fluorescence intensity of this label in concentric circles surrounding the centrosome. The radius was defined based on the point where at least 70% of the fluorescence intensity of the label is maintained (Figure 3B). Using this approach one can quantify the decrease in actin density at the centrosome upon B cell activation, as shown in Figure 3C.
To obtain greater resolution in the distribution of organelles at the IS interface, we activated B cells on antigen-coated coverslips, labeling actin, the Golgi apparatus and endoplasmic reticulum (Figure 4A). The distribution of organelles at the IS can be performed by diving the synaptic area in a central and peripheral area, applying a criteria shown in Figure 4B. This was based on previous work showing that BCRs gather within this central area, which most likely corresponds to the central supramolecular activation complex (cSMAC)10,19. Figure 4A shows that organelles recruited to the IS, such as the Golgi apparatus and the endoplasmic reticulum display opposite distributions. Indeed, their distribution indexes correlate with their immunofluorescence staining shown in Figure 4C. The Golgi apparatus shows a positive value, which means that it is concentrated at the center of the IS while the endoplasmic reticulum shows negative values, which means it is mainly localized at the peripheral region of the IS. Additionally, it is possible to take the values of the synaptic area previously determined, to measure the spreading area during B cell activation, as it is shown in Figures 4D and 4E. These results show an increase in the spreading area upon B cell activation, as previously described10,20.
As in the bead assay, we can determine the distribution of organelles toward the immune synapse by taking the XZ images of B cells activated on antigen-coated coverslips and measuring the organelle fluorescence across the Z dimension (Figure 4F). The fluorescence intensity of actin in the Z plane was measured, as it is represented in the XZ plane in Figure 4F, where a progressive enrichment of actin at the synaptic interface (fractions 1 and 2) can be observed (Figure 4G).
In addition to the B cell imaging analysis, we performed the isolation of centrosome-enriched fractions to quantify centrosome-associated proteins (Figure 5A). This approach can be important to complement the study of IS formation in B cells, given that the centrosome polarizes to the synaptic membrane and has been shown to coordinate lysosome trafficking involved in antigen extraction and presentation21. This method was previously described using large amounts of cells (1 x 109 cells)17,18 but we have standardized a simplified version, which uses a reduced amount of cells and can be performed using lower-volume ultracentrifuge-tubes (2-3 mL). As we show in Figure 5B, we analyzed different cell fractions by immunoblotting and determined which ones correspond to centrosome-rich fractions, by gamma tubulin staining. Here we show an example of proteins that undergo changes in their accumulation at the centrosome, such as Actin and Arp2, which have been previously reported5.
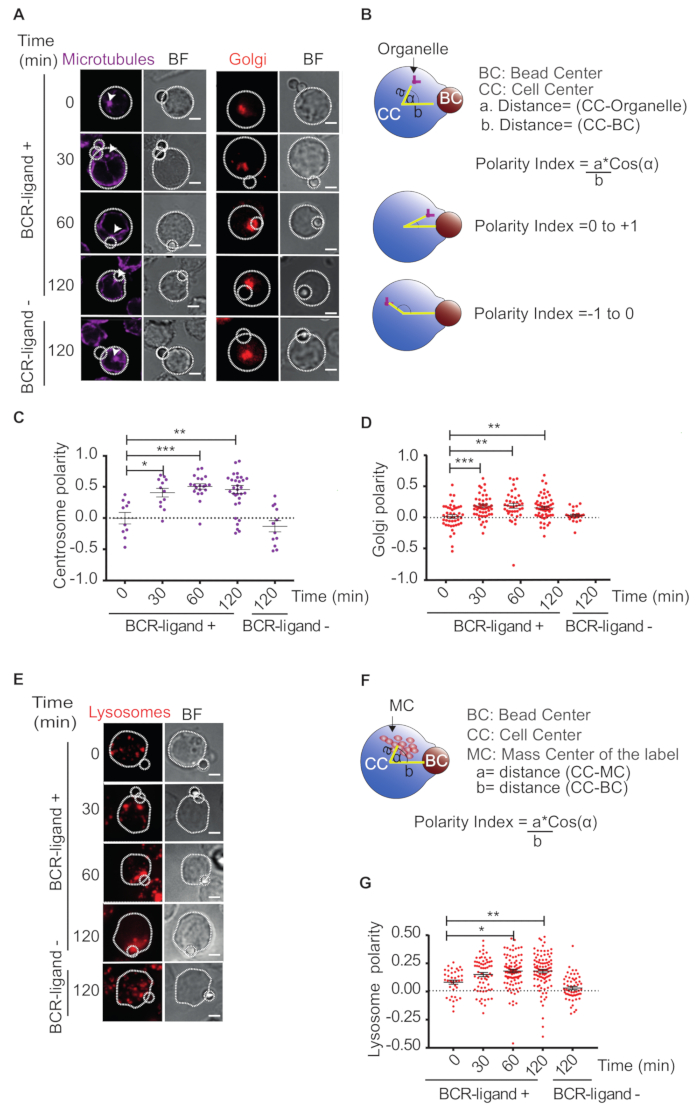
Figure 1: Polarization of organelles towards the IS. (A) Immunofluorescence staining of the Golgi apparatus (Rab6a) and microtubules (α-tubulin) in IIA1.6 B cells incubated with BCR-ligand+ (0, 30, 60 and 120 min) or BCR-ligand- beads (120 min). Arrowheads indicate the centrosome. BF = Bright field. Scale bar = 3 µm. (B) Scheme depicting how to calculate the polarity index of organelles (centrosome or Golgi apparatus) toward the IS. a = Distance between the center of the cell (CC) to the organelle. b = Distance from the CC and to the bead center (BC). (C, D) Representative dot plots showing polarity indexes of organelles at different time points of activation. Each dot represents one cell. (E) Immunofluorescence staining of Lysosomes (Lamp1) in B cells incubated with BCR-ligand+ (0, 30, 60 and 120 min) or BCR-ligand- beads (120 min). BF = Bright field. Scale bar = 3 µm. (F)Scheme representing how to calculate the polarity index of Lysosomes. a = Distance between CC to the mass center (MC). b = Distance from CC to BC. (G)Representative dot plots showing polarity indexes of lysosomes at different time points of activation. Each dot represents one cell. 2-way ANOVA with Sidak's post-test was performed. Means with SEM are shown. Please click here to view a larger version of this figure.
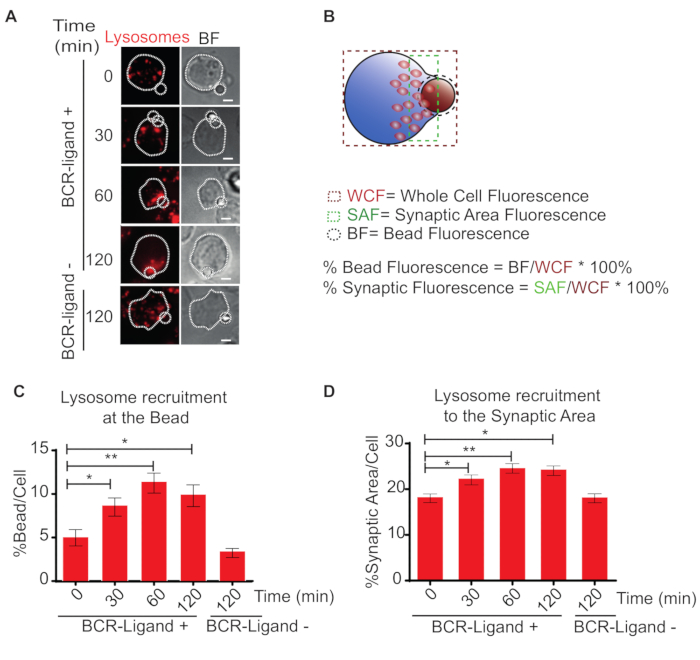
Figure 2: Accumulation of lysosomes at the IS. (A) Immunofluorescence staining of lysosomes (Lamp1) in B cells incubated with BCR-ligand+ (0, 30, 60 and 120 min) or BCR-ligand-beads (120 min). BF = bright field. Scale bar = 3 µm. (B) Scheme depicting how to calculate the accumulation of organelles such as lysosomes at the bead and the synaptic area. Fluorescence of the whole cell (WCF), bead fluorescence (BF) and the synaptic area fluorescence (SAF) are indicated. (C, D) Representative bar graphs for the accumulation of lysosomes at the bead and the synaptic area. N = 1. 20 > Cells. 2-way ANOVA with Sidak's post-test was performed. Means with SEM are shown. Please click here to view a larger version of this figure.
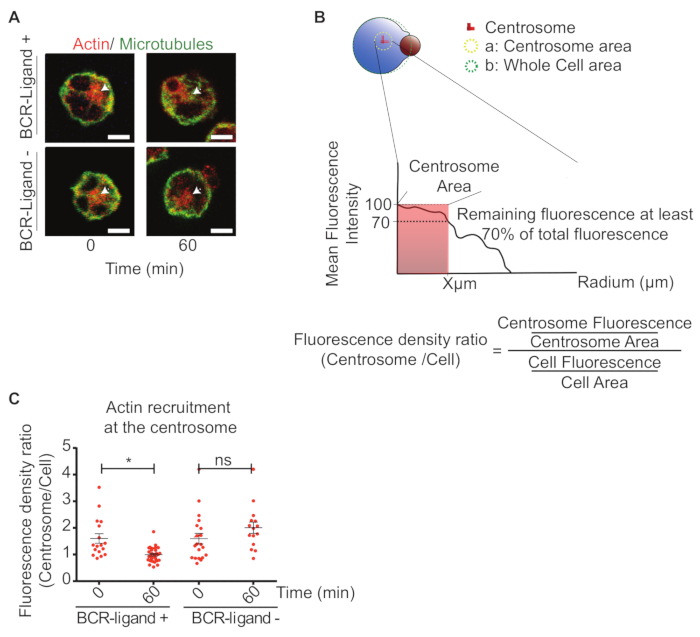
Figure 3: Quantification of actin at the centrosome during IS formation. (A) Immunofluorescence staining of actin (Phalloidin) and microtubules (α-Tubulin) in B cells incubated with BCR-ligand+ (0 and 60 min) or BCR-ligand- beads (0 and 60 min). Arrow heads indicate the centrosome. Scale bar = 3 µm. (B) Scheme depicting how to calculate the recruitment or depletion of Centrosome associated components. The centrosome area is calculated by using a radius (X µm) that maintain at least 70% of the maximum fluorescence. (C) Representative dot plots showing actin at the centrosome under activating (BCR-ligand+) and non-activating (BCR-ligand-) conditions. N = 1. 15 > Cells. 2-way ANOVA with Sidak's post-test was performed. Means with SEM are shown. Please click here to view a larger version of this figure.
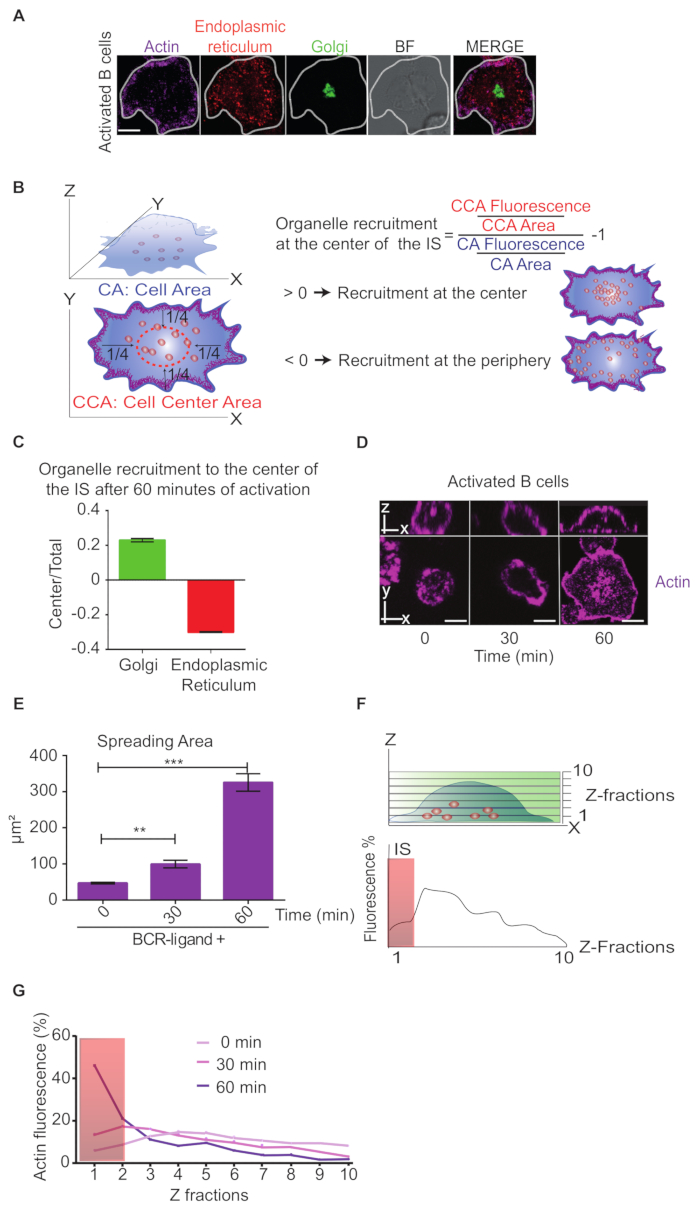
Figure 4: Distribution of cellular components at the synaptic interface. (A) Immunofluorescence staining of actin (Phalloidin), Endoplasmic Reticulum (Sec61a), Golgi apparatus (transfected with a KDELR-DN-GFP a Negative Dominant of KDEL Receptor, which localizes in the Golgi). B cells were seeded onto coverslips coated with a BCR-ligand+ for 60 min. BF = bright field. Scale bar = 5 µm. (B) Scheme depicting how to determine the spreading area of the cell and the recruitment of organelles at the center of the IS. In the scheme, CA indicates the cell area delimited by cortical actin and CCA indicates the center cell area.(C) Golgi apparatus and endoplasmic reticulum distribution at the IS, represented by a bar graph where a value >0 indicates an enrichment in the center of the IS and < 0 indicates a peripheral distribution. N = 1. 20 > Cells. Means with SEM are shown. (D) Representative images of B cells labelled for actin (Phalloidin) activated onto Ag-coated coverslips at different time points (0, 30 and 60 min), showing the XZ and XY plane. (E) Graph showing the spreading area of B cells in E. (F) Scheme depicting how to determine the distribution of the signal of interest in the Z plane, and the plot per Z fraction. The rectangle indicates the IS interface. (G) Distribution of actin across the Z plane represented by a line plot of the percentage of fluorescence distribution versus each Z fraction. The rectangle represents the IS interface. N = 1. 25 > Cells. Means with SEM are shown. Please click here to view a larger version of this figure.
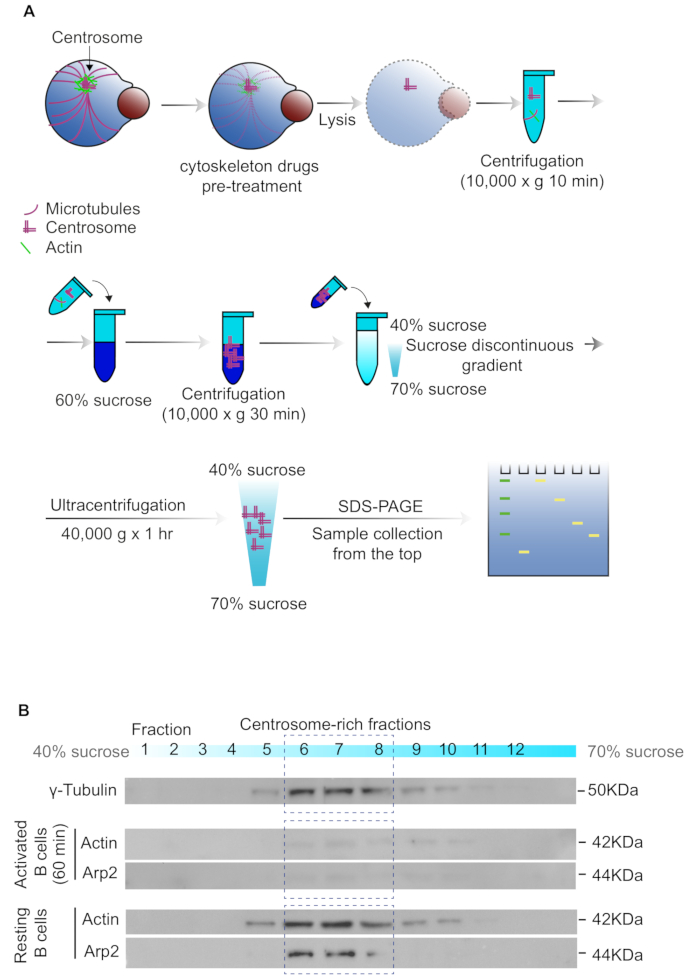
Figure 5: Centrosome enriched fractions of B cells. (A) Workflow of how to obtain centrosome-enriched fractions by sucrose gradient ultracentrifugation. (B) Immunoblot of fractions obtained from resting and activated B cells (60 min). Centrosome-rich fractions are detected by labeling with γ-tubulin and are indicated within the dashed rectangle (fraction 6 to 8). Centrosome associated proteins (Actin and Arp2) are shown in centrosome-rich fractions in activating and resting conditions. Please click here to view a larger version of this figure.
