1. Design of fluorescence in situ (FISH) anti-sense oligonucleotides to detect a specific herpesviral transcript
- Select 25 to 40 nt segments from the sequence of RNA of interest and convert to be anti-sense. A successful FISH strategy may contain from one up to ten or more different anti-sense oligonucleotides. When selecting sequences, consider the following:
- If the RNA of interest contains a unique repeat region, then capitalize on this feature and design an anti-sense oligonucleotide to target the repeat sequence.
NOTE: Tycowski and colleagues5 provide an example of this strategy with rhesus rhadinovirus (RRV) polyadenylated nuclear (PAN) RNA. - If the RNA of interest contains a known protein-binding site or stem-loop structure, design oligonucleotides that avoid these regions.
- Depending on the goals of the experiments, consider the intronic sequence and whether or not to design an anti-sense oligonucleotide for it.
- If the RNA of interest contains a unique repeat region, then capitalize on this feature and design an anti-sense oligonucleotide to target the repeat sequence.
- Perform simple computational analyses on the selected anti-sense sequences to ensure binding specificity and reduce aggregation of the anti-sense oligonucleotide.
- The sequences must be approximately 50% GC-rich (high guanine and cytosine content) and have a melting temperature in the range of 60 to 70 °C.
- Use a sequence analyzer tool to select sequences that do not self-dimerize or form hairpins with melting temperatures above 37 °C, the hybridization temperature.
- Perform a NCBI BLASTn (National Center for Biotechnology Information Basic Local Alignment Search Tool for nucleotide alignments) search of the selected sequences against both the host and viral transcriptomes using the 'somewhat similar' setting. This search will identify unique anti-sense oligonucleotides that will not likely bind to other host or viral transcripts.
NOTE: If transcriptomes are not available, perform the BLAST search with the genomic sequences. It is ideal if the searches are performed on the sequences from the virus isolated from the infected cells used in the experiment because wild strains tend to diversify and contain a combination of sequences from different lab strains.
- Order purified DNA oligonucleotides corresponding to the anti-sense sequence that has been verified computationally to be unique and likely to bind to the target RNA. No special modifications need to be introduced into the oligonucleotides.
- Test the designed anti-sense oligonucleotides for binding specificity by FISH and Northern blot.
- Using an uninfected cell-line from the same host species (e.g., 293T) and ideally the same cell type, conduct a transfection with a plasmid expressing the RNA of interest from a robust promoter (CMV, cytomegalovirus) and one with the empty vector (e.g., pcDNA3). Use a positive control for transfection such as co-transfection with a GFP (green fluorescent protein) plasmid (e.g., pmaxGFP) or the vector containing GFP.
NOTE: It is important to avoid cell-lines that have been immortalized using the herpesvirus Epstein-Barr virus (EBV) since there are sequence similarities between herpesviruses. - Perform FISH as described in section 3 on both sets of cells with the anti-sense oligonucleotides as described in this protocol. Deduce the successful candidates by comparing FISH experiments with individual, pairs, or sets of the anti-sense oligonucleotides. Use a positive control for the FISH protocol such U2 snRNA (small nuclear RNA) FISH, present at 500,000 copies per human cell nucleus6 (Table 1).
- The fluorescent signal should be specific and strong in the cell containing the RNA of interest. Design additional anti-sense oligonucleotides to strengthen the signal and remove anti-sense oligonucleotides that bind nonspecifically from consideration. Signal strength must be above background and autofluorescence.
- Test binding specificity by Northern blot.
- Using an uninfected cell-line from the same host species (e.g., 293T) and ideally the same cell type, conduct a transfection with a plasmid expressing the RNA of interest from a robust promoter (CMV, cytomegalovirus) and one with the empty vector (e.g., pcDNA3). Use a positive control for transfection such as co-transfection with a GFP (green fluorescent protein) plasmid (e.g., pmaxGFP) or the vector containing GFP.
2. Oligonucleotide and cell preparation
- Following the manufacturer's instructions, use terminal transferase to label the anti-sense oligonucleotides with dioxigenin(DIG)-dUTP or, if strongly binding, directly with a fluorescent nucleotide like Alexa Fluor 594-5-dUTP. After labeling, additional purification is not necessary. Store labeled oligonucleotides at -20 °C up to several years and in tin foil if directly labeled to prevent photobleaching.
CAUTION: The labeling solution contains a toxic material, potassium cacodylate. Handle labeling reactions with gloves.
NOTE: To conserve resources, several different anti-sense oligonucleotides can be labeled in one reaction. This protocol utilizes 3'-end labeling. Internal labeling is challenging because the chemical group (e.g., DIG or the fluorophore) gets caught or is unable to enter the active site of a DNA polymerase. An experiment with two different RNAs can be performed using directly-labeled anti-sense oligonucleotides and anti-DIG immunofluorescence with a different fluorophore (e.g., FITC (fluorescein) or Alexa Fluor 488 with Alexa Fluor 594). - Adhere the cells to the eight-chamber slides.
NOTE: Eight-chamber slides allow several simultaneous experiments while minimizing precious resources like antibodies. An alternative to eight-chamber slides is a six-well tissue culture plate with standard coverslips (22 mm x 22 mm) that are both sterile. A similar arrangement is possible with circular coverslips and a 24-well tissue culture plate. For both, increase the volumes mentioned in this protocol by 10-15x (e.g., 1.75 mL hybridization solution), and 4x (e.g., 600 µL hybridization solution) respectively.- For adherent lytic cells, use 1x trypsin/PBS at 37 °C and 5% CO2 for 10 min to suspend cells and dilute to 60% confluency.
NOTE: Adherent cell lines used in FISH experiments included 293T, iSLK.2197, and iSLK-BAC36 cells8. - Apply 200 µL of cell suspension to each chamber of the sterile eight-chambered slides and allow seed growth for 12-24 h at 37 °C and 5% CO2. Adjust as necessary for slow- or fast-growing cells and for cells that are easily damaged by trypsin.
NOTE: The objective is to have evenly spaced cells firmly attached to the slide. Consider inducing lytic phase after adhesion if the lytic cells are fragile. Conclusions drawn from experiments with iSLK cells are limited9. - For lytic suspension-cells, pre-treat eight-chamber slides with 1:10 poly L-lysine for 5 min under the tissue culture hood. Then leave the slides to dry overnight at room temperature or 1 h at 65 °C. Incubate 800 µL of lytic cells at a concentration of 1 x 106 cells/mL with the chambered slides for 30 min to 1 h at 37 °C and 5% CO2.
NOTE: Suspension-cells will settle in a monolayer, sticking to the poly L-lysine and thus excess cells are not a concern in comparison to adherent cells. Lytic cells that form grape clusters, if possible, should be separated by gentle vortexing or chemical means. Admittedly, the authors have not had much success with such recommendations in the case of lytic BJAB-RRV-GFP cells. If suspension-cells do not adhere well, consider increasing either the time or concentration of the poly L-lysine incubation.
- For adherent lytic cells, use 1x trypsin/PBS at 37 °C and 5% CO2 for 10 min to suspend cells and dilute to 60% confluency.
3. Fixation, Immunofluorescence (Optional), Hybridization, and Visualization of Viral RNAs
- Remove media and excess cells. Throughout this protocol, use vacuum suction to remove solutions and gentle micropipetting to add solutions.
NOTE: The strength of a vacuum can be reduced by placing a 200 µL micropipette tip over the glass Pasteur pipette. Replace the micropipette tip between wash steps to prevent contamination. Each wash step must be performed quickly because it is imperative that the cells never dry out. - Immediately, fix the cells with pre-chilled 4% formaldehyde/PBS (phosphate-buffered saline) on ice for 30 min. Wash the cells three times with 200 µL 1x PBS cooled to 4 °C and incubate for 5 min at room temperature or on ice.
- Permeabilize the fixed cells with 200 µL of pre-chilled 0.5% Triton-X/PBS (phosphate buffered saline) for 10 min on ice or 750 µL of pre-chilled 70% ethanol at 4 °C for 1 h (min) to 7 d (max).
NOTE: Collect protein, total RNA, and genomic DNA samples at the point of fixation to ensure consistency between images and biochemical assays. All washes throughout this protocol are performed in the same manner unless otherwise specified. 70% ethanol loosens the glue between the chambers and the slide, which eases later separation, and also provides a significant pause in the protocol. Nonetheless, use paraffin film around the chamber slide to reduce evaporation and check the level of the ethanol in each chamber about every 8 h. 70% ethanol also flattens the cells, making a crisper image, while Triton-X does not dehydrate the cells and change the dimensions of the cell.
- Permeabilize the fixed cells with 200 µL of pre-chilled 0.5% Triton-X/PBS (phosphate buffered saline) for 10 min on ice or 750 µL of pre-chilled 70% ethanol at 4 °C for 1 h (min) to 7 d (max).
- Remove chambers carefully to prevent cracking the slide. If the experiment includes immunofluorescence (IF) of a viral or host protein with a polyclonal primary antibody, perform the IF as described below before proceeding to RNA FISH. If the immunofluorescence uses a monoclonal primary antibody, then perform immunofluorescence as described in step 3.3.1 after step 3.11.
NOTE: Use a fresh removal device or one with very little leftover adhesive provided by the manufacturer and gently ease the chambers off to prevent the slide from cracking. Using 70% ethanol as the permeabilizing reagent for 4 h greatly reduces the likelihood of cracking. In the case of a crack, continue the protocol on chambers not affected by the crack and be mindful of the higher oxidation rate of imperfectly sealed slides (i.e. decreased storage life).- Rinse cells with pre-chilled 1x PBS and block with pre-chilled 4% BSA (bovine serum albumin)/1x PBS for 30 min at 4 °C.
NOTE: The use of BSA throughout this protocol limits nonspecific labeling. - Remove blocking solution and incubate the cells with 1:200 or another polyclonal primary antibody in 0.1% BSA/1x PBS for 1 h at 4 °C. Then wash three times with 1x PBS.
NOTE: An antibody10 for detection of SSB/ORF6 (viral single-stranded DNA binding protein) was used at 1:200 dilution. - Incubate the cells with a secondary antibody with fluorophore compatible with the FISH-detecting antibody for 1 h at 4 °C. Wash three times with 1x PBS. Then fix with 4% formaldehyde/1x PBS for 10-15 min and permeabilize with either Triton-X or 70% ethanol as previously described before proceeding to FISH. Cover slide with tin foil to preserve fluorescent signal and prevent photobleaching.
- Rinse cells with pre-chilled 1x PBS and block with pre-chilled 4% BSA (bovine serum albumin)/1x PBS for 30 min at 4 °C.
- Wash the cells with 2x SSC (saline sodium citrate) once and then apply 45 µL of hybridization solution consisting of 50% formamide, 10% dextran sulfate, 2x SSC, 0.1% BSA, 500 µg/mL salmon sperm DNA, 125 µg/mL E. coli tRNA, and 1 mM vanadyl ribonucleoside complexes. Incubate for 1 h at 37 °C in a humidity chamber that can be a 150 mm Petri dish with moistened sterile wipes.
NOTE: Prepare fresh hybridization solution at least an hour before use. Dissolve the dextran sulfate in water first, vortexing frequently and incubating in a 37 °C water bath. - Calculate to have a suggested concentration of 25 µM oligonucleotides in 35-uL hybridization solution per chamber. Adjust concentration of anti-sense oligonucleotide as needed. Add distilled water to the oligonucleotides to bring the denaturation volume to 10 µL.
NOTE: Following the labeling reaction, the oligonucleotides are stored in the quenched solution containing 0.18 M potassium cacodylate, 23 mM Tris-HCl, 0.23 mg/mL BSA, 4.5 mM CoCl2, 18 mM EDTA, 2.7 mM K-phosphate, and 6.8 mM KCl, 45 µM 2-Mercaptoethanol, 0.02% Triton X-100, and 2% glycerol. The concentrations are high enough that dilution with water will bring the denaturation solution to concentrations near to 1x TE (10 mM Tris-HCl and 1 mM EDTA), a standard oligonucleotide denaturation buffer. - Denature the DIG- and/or Alexa Fluor 594-labeled oligonucleotides at 95 °C for 5 min. Then add 35 µL fresh hybridization solution per intended chamber to the denatured oligonucleotides. If performing double FISH, both sets of anti-sense oligonucleotides may be denatured and hybridized together.
- Remove the pre-hybridization solution and then add hybridization solution containing the labeled oligonucleotides to the cells. Incubate overnight in the humidity chamber at 37 °C with tin foil to protect the fluorophore-labeled oligonucleotides.
NOTE: Incubation should be at least 10 h and not more than 24 h. - The next day, wash the cells twice with 2x SSC for 10 min at 37 °C and then twice with 1x SSC for 10 min at 25 °C.
- Fix the cells with pre-chilled 4% formaldehyde/1x PBS for 10-15 min on ice. Then wash the cells with PBS three times and permeabilize for 1 h with pre-chilled 70% ethanol or for 10 min with pre-chilled 0.5% Triton-X/1x PBS at 4 °C.
- Incubate the cells with 1:200 anti-DIG FITC in pre-chilled 0.1% BSA/1x PBS for 1 h at 4 °C. Remove the antibody solution and wash three times with 1x PBS.
- Fix with pre-chilled 4% formaldehyde/1x PBS for 10-15 min at 4 °C and then wash three times with 1x PBS. If performing immunofluorescence for a host or viral protein with a monoclonal primary antibody, permeabilize the cells and then perform the IF protocol outlined in step 3.3.1. Otherwise proceed to the DAPI staining.
- Incubate the cells with 0.4 µg/mL DAPI in pre-chilled 0.5% Triton-X/1x PBS for 15 min on ice and then wash three times with 1x PBS.
- Mount slides with fluorescent beads (optional) and a mounting medium. Then seal the coverslip to the slide with clear nail polish.
- Using a confocal microscope, collect images of the samples within an hour to a week of performing the protocol at 630x magnification. Apply multiple coats of nail polish to seal the cover slip and to prolong fluorophore life by reducing the rate of oxidation.
NOTE: Do not use a DAPI-containing mounting medium. When collecting the images, include the scale bar on each image for later quantification. Fluorescent beads serve as controls of fluorescence intensity between slides and sample preparations11. Acquire images at the midsection of the cell for two-dimensional (2D) quantification in step 4.
4. Quantification of FISH and IF images to highlight subcellular localization and to determine nucleocytoplasmic ratio of fluorescence
- Perform image analysis on an assembled stack of the various fluorescent-stained and merged images to ensure consistency. Set the scale of the image analysis software using the scale bar included when the images were collected.
- To quantify fluorescence intensity across several channels and in reference to the nuclear DAPI stain, use a line tool and a plot-profile function. Then indicate the line permanently on a copy of the image using markers that do not obstruct or influence the viewer's judgment.
- Establish criteria to guide where the line is drawn such as a trace that captures a diversity of topographical features, peaks and valleys, along a central axis or a line that does not traverse supersaturated areas.
NOTE: These line traces depict raw fluorescence in a cell and thus are limited to comparisons of the locations of a stain, not intensity. To compare intensities of the same stain between slides, treatments, or preparations, add a fluorescent bead to the slide as an internal control during step 3.13. The fluorescent bead must be added during the mounting process and detected with the same settings on the excitation laser and photomultiplier tube (confocal).
- Establish criteria to guide where the line is drawn such as a trace that captures a diversity of topographical features, peaks and valleys, along a central axis or a line that does not traverse supersaturated areas.
- To quantify a shift in subcellular localization, calculate nucleocytoplasmic ratios of cells undergoing different treatments.
- Measure the area and raw fluorescence intensity of both the nucleus and cytoplasm using the nuclear DAPI stain to set the inner boundary. Include nuclear and cytoplasmic controls such as a nuclear RNA (e.g., KSHV PAN RNA) and cytoplasmic RNA (e.g., host GAPDH mRNA). Moreover, calculate background intensity for three cell-like areas and average the values per pixel or µm2.
NOTE: Intensity values tend to lack units and so the term 'units' is used. - Normalize both nuclear and cellular raw intensity values by first determining the average background for the same area and then subtracting that individualized value from the raw intensity of the area.
- For example, a nucleus of a lytic B-cell has an area of 133.4 µm2 and a raw intensity of 75976 units while the background intensity for the same fluorescent signal was determined to be 0.67 units per µm2. The normalized nuclear intensity would be
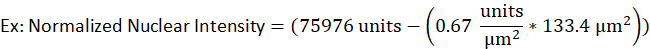
- For example, a nucleus of a lytic B-cell has an area of 133.4 µm2 and a raw intensity of 75976 units while the background intensity for the same fluorescent signal was determined to be 0.67 units per µm2. The normalized nuclear intensity would be
- Enter the values in the following equation.

NOTE: This calculation controls for changes in subcellular area. Lytic induction and drug treatments can enlarge the nucleus or change the size of the cell, respectively. - To interpret the results, create a box whisker plot. An equal distribution of the fluorescent signal would be close to zero, whereas a nuclear distribution would favor a positive ratio value and a cytoplasmic distribution would trend toward a negative ratio value.
- Measure the area and raw fluorescence intensity of both the nucleus and cytoplasm using the nuclear DAPI stain to set the inner boundary. Include nuclear and cytoplasmic controls such as a nuclear RNA (e.g., KSHV PAN RNA) and cytoplasmic RNA (e.g., host GAPDH mRNA). Moreover, calculate background intensity for three cell-like areas and average the values per pixel or µm2.
The FISH and IF methods detailed in this manuscript are shown in Figure 1 along with the quantification of results by line traces of fluorescent intensity. The results presented here are semi-quantitative and offer insight into localization, rather than into comparisons between intensities of different fluorescent stains because experiments did not include a fluorescent bead in the slide preparation. Figure 1 also reveals that the cytoplasmic and nuclear areas and their ratios are different for latent and lytic KSHV-infected cells. Thus, area is controlled in the nucleocytoplasmic ratio showcased in Figure 2. Figure 2 validates the calculation detailed in this manuscript for a nucleocytoplasmic ratio with the use of a nuclear control, the viral polyadenylated nuclear (PAN) RNA, and a cytoplasmic control, the host GAPDH mRNA. Figure 3 reveals that when KSHV DNA replication is inhibited in the lytic phase by the use of either phosphonoacetic acid (Doxy + PAA) or cidofovir (Doxy + Cido), the early ORF59-58 transcript shifts to a predominantly cytoplasmic localization. The micrographs and the two quantification methods in Figure 3 support this result and reveal that PAN RNA localizes to specific nuclear sites despite inhibition of viral DNA replication and the change seen for early ORF59-58 transcript.
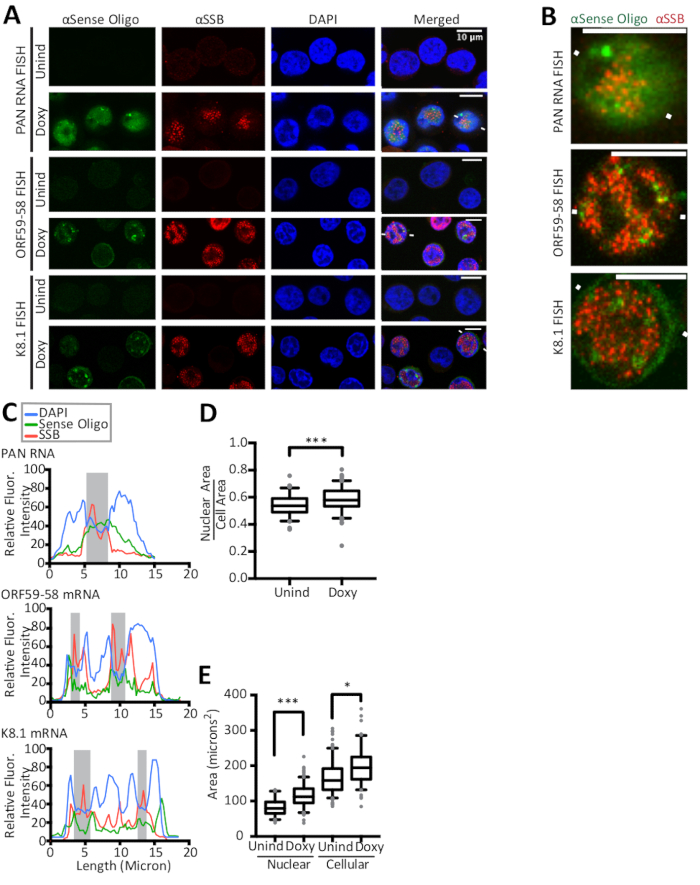
Figure 1: Lines traces of fluorescent intensity reveal subtleties in fluorescence in situ hybridization (FISH) of KSHV transcripts and immunofluorescence (IF) of KSHV replication compartments. (A–B) Confocal images of TREx RTA (tetracycline inducible viral replication and transcription activator protein) BCBL-1 cells12 that have been induced into the lytic phase for 24 h with doxycycline (Doxy). Scale bar indicates 10 µm. (A) Fluorescence in situ hybridization (FISH) for viral RNAs (green) and immunofluorescence (IF) for viral single-stranded DNA binding protein (ORF6/SSB) (red), a component of KSHV replications compartments, reveal that viral transcripts localize in the cytoplasm, nucleus, and in nuclear foci outside ORF6/SSB enriched areas, also known as replication compartments. The anti-SSB antibody10 was diluted to 1:200 in 0.4% BSA/1x PBS and detected with 1:500 anti-rabbit Alexa Fluor 594 secondary antibody in 0.4% BSA/1x PBS. All anti-sense oligonucleotides used throughout this study are provided in Table 1. The detection of ORF59-58 mRNA includes both the bicistronic and monocistronic transcripts. However, in KSHV-infected JSC-1 cells, the monocistronic mRNA is at least 18-fold less abundant than the bicistronic transcript and likely contributes only a minor portion of the total fluorescent signal observed13. Moreover one of the PAN RNA oligonucleotides (SB88) can also detect the viral transcript for K7. The signal from a detection of K7 will not be as significant compared to the signal detecting KSHV PAN RNA, which is present at nearly 80% of all polyadenylated RNA in a lytic KSHV-infected cell14. Additionally one of the four anti-sense oligonucleotides (tkv13) in the detection of the K8.1 mRNA is able to bind to multiple isoforms of K8.1 and other isoforms of nearby open reading frames (ORF). The FISH signal from only oligonucleotide tkv13 is insufficient (data not shown). The combined hybridization of the four oligonucleotides and the binding of them on the same transcript likely provides the observed strong signal. White lines flanking cells in (A) depict the line path of fluorescence intensities for the FISH and IF signals, plotted in (C). (B) Digitally zoomed images of cells in (A) flanked by white lines. For simplicity, the blue DAPI channel is omitted. (C) The plots show the relative fluorescent intensities for each stain along the same line: αSSB (red), viral transcripts (green; transcript indicated on plot), and DAPI (blue). Shaded areas indicate DAPI-reduced regions that correspond to viral replication compartments or SSB/ORF6-enriched areas. (D) The ratio of nuclear area to cellular area changes and thus the fluorescence intensity ratio used throughout was normalized for area. (E) Nuclear and cellular areas measured for TREx RTA BCBL-1 cells with and without undergoing lytic activation. Statistically significant changes are seen compared to uninduced cells. The box and whisker plots represent the 10 and 90 percentiles. Figure reprinted with slight modifications from Vallery, Withers, and colleagues15 under a Creative Commons Attribution license. Please click here to view a larger version of this figure.
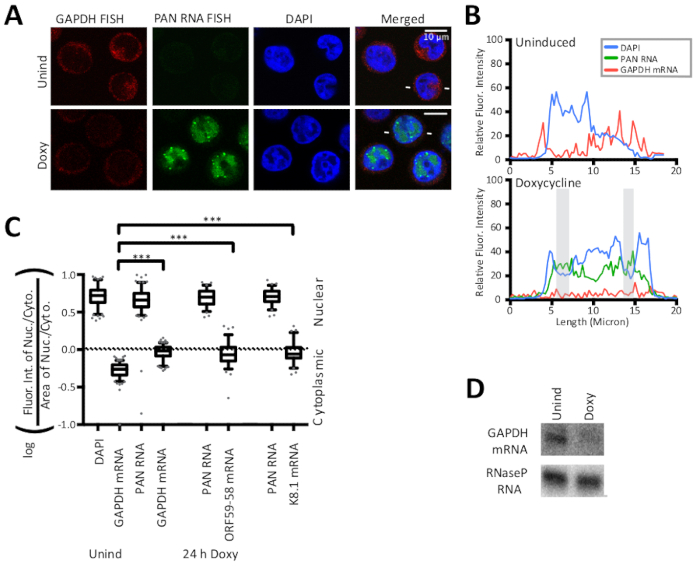
Figure 2: Control nuclear and cytoplasmic FISH strategies validate the calculation method of the nucleocytoplasmic ratio. (A) FISH for the host GAPDH mRNA (red) and for the viral polyadenylated nuclear (PAN) lncRNA (green) and DAPI nuclear staining (blue) are positive FISH controls for the calculation method determining the nucleocytoplasmic ratio. Host GAPDH mRNA is a canonical target of the KSHV's host shutoff effect and is degraded upon lytic induction as shown here. (B) Fluorescence intensities along a line indicated by white lines flanking lytic cells in (A). DAPI (blue), PAN RNA (green), and GAPDH mRNA (red). Shaded areas are as defined in Figure 1. (C) Quantification of the fluorescence intensities of cells represented by (A) (n = 150 for each GAPDH sample, n = 75 for the ORF59-58 or K8.1 samples) were performed for three biological replicates of cells shown in Figure 2 and Figure 3. P-values: >0.05 (ns), <0.05 (*), <0.005 (**), and <0.0005 (***). (D) Representative Northern blot of RNA from TREx RTA BCBL-1 cells 24 h after Doxy. The box and whisker plots represent the 10 and 90 percentile. Figure reprinted with slight modifications from Vallery, Withers, and colleagues15 under a Creative Commons Attribution license. Please click here to view a larger version of this figure.
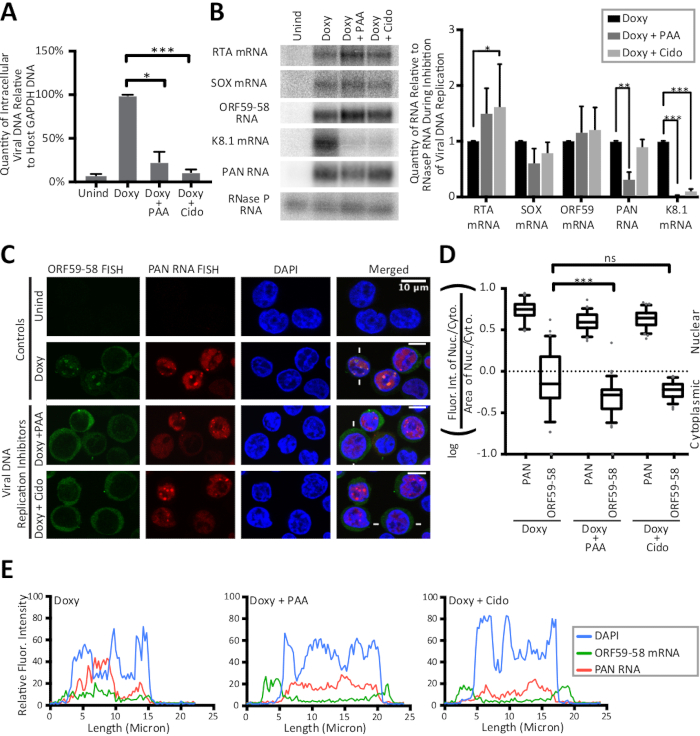
Figure 3: Line traces and calculation of nucleocytoplasmic ratios reveal a strong shift to the cytoplasm for the early lytic ORF59-58 transcript upon inhibition of viral DNA replication. TREx RTA BCBL-1 cells were treated for 24 h with no drug (Unind), doxycycline only (Doxy), or with doxycycline and one inhibitor of herpesviral DNA replication, phosphonoacetic acid (Doxy + PAA) or cidofovir (Doxy + Cido). Panels (A–C) show data from samples collected from three biological replicates. (A) qPCR values for viral intracellular DNA during inhibition of viral DNA replication were normalized to the quantity of promoter DNA of the host-cell GAPDH gene. (B) Northern blot (left) and quantification (right) show total RNA levels during inhibition of viral DNA replication. Uninduced levels of all RNAs were undetectable. (C) Representative FISH images for viral ORF59-58 transcripts (green) and PAN RNA (red) upon inhibition of viral DNA replication. DAPI (blue) was the nuclear stain. (D) Quantification of the fluorescence intensities of cells represented by (C) (n = 75 each) was done on biological triplicates. (E) Fluorescence intensities along lines drawn across cells indicated by white lines in (C) are shown: DAPI (blue), PAN RNA (red), and ORF59-58 mRNA (green). P-values: >0.05 (ns), <0.05 (*), <0.005 (**), and <0.0005 (***). The sequences of all oligonucleotides in this study are provided in Table 1. The box and whisker plots represent the 10 and 90 percentiles. Figure reprinted from Vallery, Withers, and colleagues15 under a Creative Commons Attribution license. Please click here to view a larger version of this figure.
| Northern Oligos | |||||||
| Oligo No. | Gene | Sequence | Position within the gene | Position with the Reference NC009333.1 Genome (Numbers do not reflect direction or strand) | |||
| KORF50 | KSHV RTA/ORF50 | CGCATTGCGGTGGTTGAAATTGCTGG | 1284 to 1309 | 73936 to 73961 | |||
| JBW249 | KSHV SOX/ORF37 | TAACCTGACACCACCAAACACACGGTCCAC | 262 to 291 | 57633 to 57662 | |||
| tkv379 | KSHV ORF59-58 | TGGAGTCCGGTATAGAATCGGGAACCT | 941 to 967 (ORF59 ORF) | 95879 to 95905 | |||
| tkv13 | KSHV K8.1 | AAGGCATAGGATTAGGAGCGCCACAGGGATTTCTGTGCGAAT | 16 to 57 | 76029 to 76070 | |||
| SB2 | KSHV PAN RNA | ACAAATGCCACCTCACTTTGTCGC | 664 to 687 | 29496 to 29519 | |||
| Rnase P | Human RNase P | TGGGCGGAGGAGAGTAGTCTG | 319 to 339 | N/A | |||
| FISH Probes | |||||||
| Oligo No. | Gene | Sequence | |||||
| SB2 | KSHV PAN RNA | ACAAATGCCACCTCACTTTGTCGC | 664 to 687 | 29496 to 29519 | |||
| SB85 | KSHV PAN RNA | CGCTGCTTTCCTTTCACATT | 373 to 392 | 29205 to 29224 | |||
| SB88 | KSHV PAN RNA | GTGAAGCGGCAGCCAAGGTGACTGG | 1 to 22 | 28830 to 28854 | |||
| tkv13 | KSHV K8.1 | AAGGCATAGGATTAGGAGCGCCACAGGGATTTCTGTGCGAAT | 16 to 57 | 76029 to 76070 | |||
| tkv14 | KSHV K8.1 | TGATATTAAGGCATCGGTCAGTTCTGTGGTGGCCTGGA | 377 to 414 | 76390 to 76427 | |||
| tkv15 | KSHV K8.1 | GTAAGGTTACGCTTTATCCCTACACACCGACGGTTTACCC | 461 to 500 | 76474 to 76513 | |||
| tkv16 | KSHV K8.1 | GGACAAGTCCCAGCAATAAACCCACAGCCCATAGTATG | 688 to 725 | 76701 to 76738 | |||
| tkv376 | KSHV ORF59-58 | TAATGTGTTCATTGACCCTCCTGATT | 54 to 79 | 96767 to 96792 | |||
| tkv377 | KSHV ORF59-58 | GCCGATCCGTGCACTTGCACTACTCCGGTT | 93 to 122 | 96724 to 96753 | |||
| tkv378 | KSHV ORF59-58 | AAGGCTATGCCAGCGTCGAGTACATTCGCA | 300 to 329 | 96517 to 96546 | |||
| tkv379 | KSHV ORF59-58 | TGGAGTCCGGTATAGAATCGGGAACCT | 941 to 967 | 95879 to 95905 | |||
| tkv380 | KSHV ORF59-58 | AAAGAGTGTGAACGAGTACAGGGCCTT | 1289 to 1315 | 95531 to 95557 | |||
| tkv381 | KSHV ORF59-58 | AAACACTGCTGACGCGCAGATCCATTCC | 1423 to 1450 | 95396 to 95423 | |||
| tkv382 | KSHV ORF59-58 | TACCTGTGTACTATTGGCGGCGCCTGATACAC | 1571 to 1602 | 95244 to 95275 | |||
| tkv383 | KSHV ORF59-58 | GGGTCGAGATTCAGCTAATTAGGCGAAAACTCCACAGG | 2136 to 2173 | 94673 to 94710 | |||
| Stellaris | GAPDH | Premade by Stellaris | N/A | N/A | |||
| qPCR Primers | |||||||
| tkv458 | GAPDH Promoter | CTGCACCACCAACTGCTTAG | N/A | N/A | |||
| tkv459 | GAPDH Promoter | GTCTTCTGGGTGGCAGTGAT | N/A | N/A | |||
| tkv319 | KSHV ORF39 (gM) | GTGAGGTGCTTCGCTGAGTT | N/A | 60075 to 60094 | |||
| tkv320 | KSHV ORF39 (gM) | CCTGGGTCAAGCTGTTGTTT | N/A | 60218 to 60237 | |||
| RT-qPCR Primers | |||||||
| tkv 455 | K8/K-bZIP Forward RT qPCR Primer | CGAAAGCAAGGCAGATACG | 655 to 673 | 75603 to 75621 | |||
| tkv 456 | K8/K-bZIP Reverse RT qPCR Primer for unspliced | GCCATTGTTCCCATTTGAGT | 755 to 774 | 75703 to 75722 | |||
| tkv 457 | K8/K-bZIP Reverse RT qPCR Primer for spliced | CATCAGCATGTCGCGAAG | 871 to 888 | 75819 to 75836 | |||
| JBW479 | Human RNase P Forward | AGCTTGGAACAGACTCACGG | 238 to 257 | N/A | |||
| JBW480 | Human RNase P Reverse | GCGGAGGAGAGTAGTCTGAA | 317 to 336 | N/A | |||
Table 1: All oligonucleotides used in the analyses of this publication. Table 1 was reproduced with permission from the American Society for Microbiology under a Creative Commons Attribution license from Vallery et al.15.
