Study 1
Liver was harvested from two cohorts of male Sprague Dawley rats administered corn oil for 4 days, staggered by one week. Slides were prepared from freshly minced tissue, frozen minced tissue, and frozen cubed tissue processed in Merchant’s medium or mincing solution using the tissue mincing device. Frozen tissues obtained from animals from the first cohort were evaluated after freezer storage for ~3.5 months. Frozen tissues obtained from animals from the second cohort were evaluated after storage for ~6 months. Comets exhibiting the characteristic “hedgehog” morphology, indicative of heavily damaged cells (of uncertain etiology that may include cytotoxicity or mechanical stress), were not included in the scoring of % tail DNA; the percentage of hedgehogs, identified solely upon visual inspection of morphology, was tabulated separately as per OECD TG 4899. The mean % tail DNA results for the animals in the two cohorts (based on scoring 100 cells/animal) are summarized in Figure 2A. All the methods using frozen tissue produced values higher, although not always statistically different, than fresh tissue. Considering that the recommended upper limit for % tail DNA in fresh rat liver is 6%9, these results demonstrate that any of these methods can work well for evaluating frozen tissue. Mincing solution performed at least equally as well as Merchant’s medium; since the latter is more time-consuming to prepare, mincing solution was selected for subsequent studies. There were no statistically significant differences in damage in tissues from the second animal cohort that had been stored frozen for ~6 months prior to processing as compared to the tissues from the first cohort processed after ~3.5 months of storage. Overall, the % hedgehogs paralleled the % tail DNA although the % hedgehogs in the frozen tissues relative to fresh tissue was more variable than for % tail DNA (Figure 2B). The values for these endpoints in the fresh tissue provide a baseline against which the DNA damage artefactually introduced by the freezing methods can be gauged.
Study 2
Female B6C3F1 mice were administered normal saline or the mutagen, ethyl methanesulfonate (EMS) at 200 mg/kg/day in normal saline for three days. Tissue was harvested 3 h after the final dose administration and processed fresh in the comet assay. Additional tissue was flash frozen after being minced in mincing solution or collected as cubes. Cubed samples were subsequently processed in mincing solution using the sieve device immediately prior to preparation of comet slides. On the premise that cells exhibiting the hedgehog morphology represent cells at the high end of the continuum of chemical-induced DNA damage, hedgehogs scorable by the imaging software were included in the automated scoring of % tail DNA. The % tail DNA results (based on scoring 150 cells/animal) are summarized in Figure 3. Liver tissue frozen as cubes and subsequently processed using the tissue mincing device yielded results very similar to those obtained for freshly minced tissue. The % tail DNA values were somewhat higher for the frozen minced tissue, but baseline values for the frozen minced tissue were below 6%, as recommended for fresh rat liver in the OECD test guideline for the comet assay9. A statistically significant increase in EMS-induced DNA damage was measured in tissue samples processed by all three methods.
Study 3
Portions of liver tissue collected from female B6C3F1 mice in a 90 day toxicity study of a test chemical conducted by a remote laboratory were minced or cut into cubes, flash frozen, and shipped overnight on dry ice to this laboratory for analysis. Minced tissue was analyzed following ~2 months of freezer storage (Figure 4). Cells that appeared upon visual inspection to be hedgehogs but were scorable by the imaging software were included in the % tail DNA measurements (150 cells scored/animal). The number of hedgehogs (whether scorable or nonscorable) identified solely upon visual inspection of morphology in a total of 150 cells/animal was tabulated separately to provide information on the frequency of these highly damaged cells. Overall, DNA damage (measured as % tail DNA) in frozen minced tissue was high across all the dose groups with substantial animal to animal variability, including in the vehicle control group. The % tail DNA results correlated with % hedgehogs (identified solely on the basis of morphology), indicating that hedgehogs contributed substantially to the high level of DNA damage that was observed in these samples. Based on the high background level of DNA damage measured in the minced tissue, frozen cubed tissue was subsequently analyzed after ~22 months of storage (Figure 4). Both DNA damage and % hedgehogs were very low in the flash frozen cubed tissue that had been prepared in parallel with the minced tissue samples. Thus, the highly damaged cells reflected by the hedgehog morphology in the minced tissue samples were clearly not the result of a biological process, but were likely introduced by mechanical disruption and/or tissue warming during the mincing procedure prior to being flash frozen; these data were considered to be unreliable. Fortunately, the analysis of frozen cubed tissue provided a clear result, namely the lack of a DNA damage response in the liver of female mice following three months of exposure to the test chemical. The results provided by this case study exemplify the risk associated with preparation of minced tissues by a relatively inexperienced laboratory and demonstrate the utility of the frozen cubed tissue method to circumvent this problem.

Figure 1: Tissue mincing device and plunger used to prepare a single cell suspension from frozen tissue. The prototype device (4.5 mm inner diameter, 0.4 mm mesh size) was kindly provided by Dr. Gunnar Brunborg (NorGenotech, Oslo, Norway). The panel on the right provides an example of an appropriately sized cube of tissue for use with the device. Please click here to view a larger version of this figure.
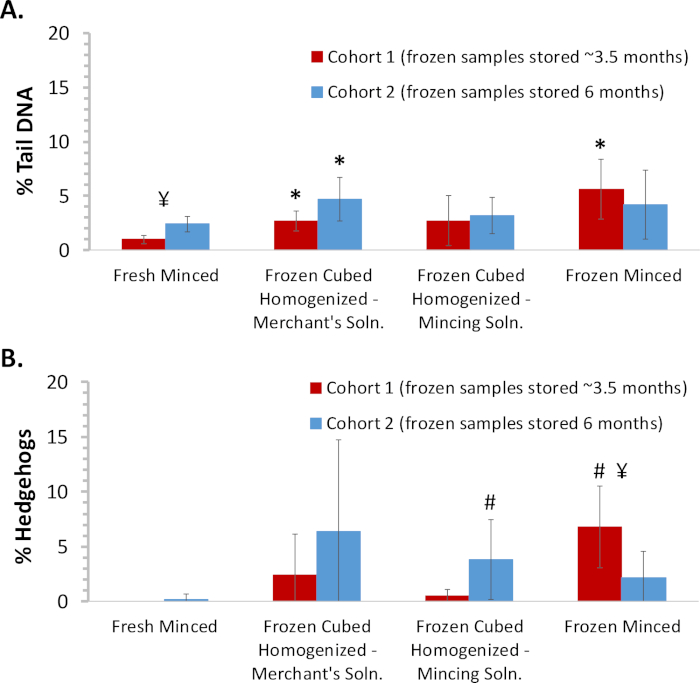
Figure 2: Comparison of DNA damage measured in fresh and frozen minced and homogenized mouse liver samples. Liver was harvested from two staggered cohorts (n = 5/group/cohort) of male Sprague Dawley rats administered corn oil for 4 days. Slides were prepared from freshly minced tissue, frozen minced tissue, and frozen cubed tissue processed in Merchant’s medium or mincing solution using the tissue mincing device. Frozen tissues were analyzed ~3.5 and 6 months following necropsy for cohorts 1 and 2, respectively. Comets classified as hedgehogs based on morphology were not included in the scoring of % tail DNA. (A) Cohort mean % tail DNA results. (B) Cohort mean % hedgehog results. Error bars reflect standard deviation. Statistical analyses were conducted to compare frozen tissues against fresh tissue for each cohort and to compare the results between the two cohorts for each tissue preparation method. *Statistically different (p < 0.05) from fresh minced tissue within the same cohort using the Student's t-test; #statistically different (p < 0.05) from fresh minced tissue within the same cohort using the Mann-Whitney test for non-normally distributed data; ¥ statistical difference (p < 0.05) between cohorts using the Student’s t-test. Please click here to view a larger version of this figure.
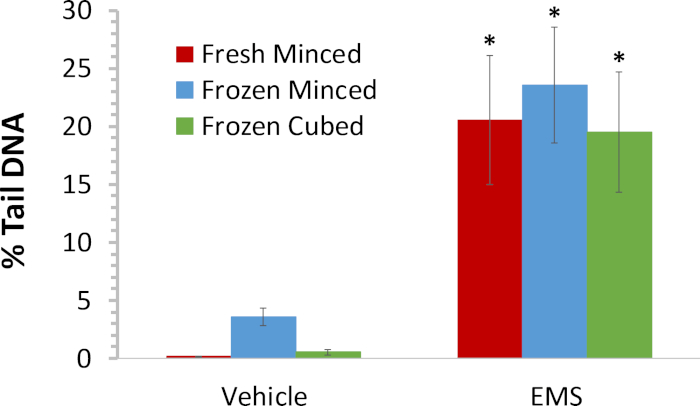
Figure 3: Comparison of DNA damage induced by EMS in Liver tissue processed by different procedures. Mean % tail DNA for female B6C3F1 mice administered vehicle or 200 mg/kg/day EMS for three days (n = 5/group). Livers were harvested 3 h following the final dose administration and processed fresh in the comet assay. Additional liver tissue was flash frozen after being minced in mincing solution or collected as cubes; cubed samples were homogenized using the tissue mincing device immediately prior to preparation of comet slides. To ensure cells at the high end of the continuum of EMS-induced DNA damage were not excluded from the analysis, comets that upon visual inspection appeared to meet the morphological description of hedgehogs but were found to be scorable by the imaging software were included. Error bars reflect standard deviation. A Student’s t-test was used to compare the amount of DNA damage in animals exposed to EMS against that in the vehicle control animals for each sample preparation method. *Statistically significant increase at p < 0.05. Please click here to view a larger version of this figure.
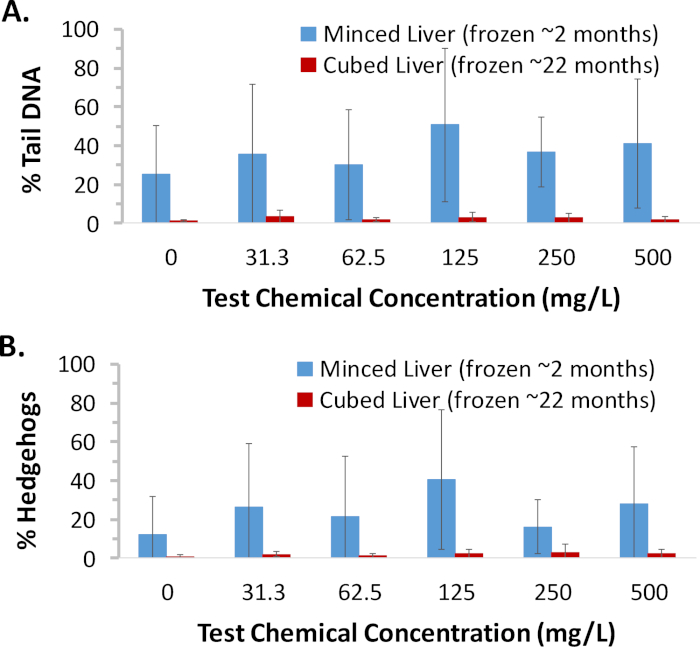
Figure 4. Case study demonstrating utility of frozen cube method and high quality data obtained after prolonged storage. Portions of liver tissue collected from female B6C3F1 mice exposed to various concentrations of a test chemical for ~90 days (n = 5/group) were minced or cut into cubes and flash frozen at the in-life laboratory and subsequently shipped to this laboratory for analysis. Minced tissue was analyzed following ~2 months of storage; due to the poor quality of the data, frozen cubed tissue was subsequently analyzed following ~22 months of storage. To ensure cells at the high end of the continuum of chemical-induced DNA damage were not excluded from the analysis, data from scorable cells that, upon visual inspection appeared to be hedgehogs, were included. (A) Group mean % tail DNA results. Compared to minced tissue, high quality results were obtained from frozen cubed tissue, even after prolonged storage. (B) Group mean % hedgehog results. Poor mincing technique is evident from the extensive non-biological DNA damage observed as hedgehog comets in the minced tissue samples. Error bars reflect standard deviation. An ANOVA with Dunnett’s test was used to evaluate for a positive response to the test chemical for each sample preparation method. There were no statistically significant (p < 0.05) dose groups detected for either method. Please click here to view a larger version of this figure.
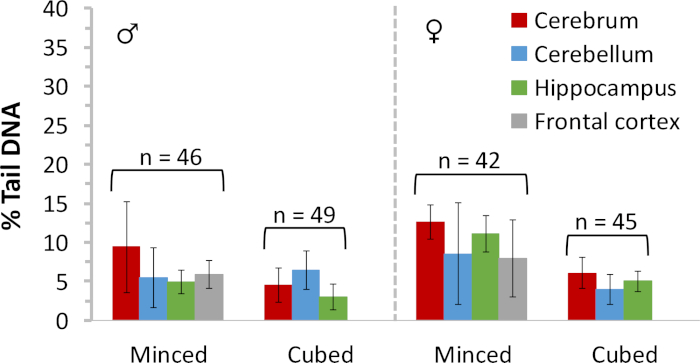
Figure 5: Baseline DNA damage measured in frozen rat brain tissues. Comet assay results are provided for frozen minced or cubed tissues representing several different regions of the brain of male and female Sprague Dawley rats. Data (generated by including scorable hedgehog comets) were collated over several studies; n = total number of brain tissues evaluated for each sample preparation method. Error bars reflect standard deviation. Please click here to view a larger version of this figure.
