1. Preparation of Culture Medium for NK Cells and Hepatic Tumor Cells
- Use human natural killer cells (e.g., NK92MI) and human liver cancer cell line (e.g., SK-HEP-1).
- Prepare NK cell culture medium for NK92MI human NK cells by adding the following components to 500 mL of minimum essential medium Eagle alpha without ribonucleosides and deoxyribonucleosides to the indicated final concentrations: 0.02 mM folic acid (100 µL of 100 mM folic acid), 0.2 mM myoinositol (500 µL of 200 mM myoinositol), 0.1 mM β-mercaptoethanol (3.5 µL of 14.3 M β-mercaptoethanol), 2 mM L-glutamine (5 mL of 200 mM L-glutamine), 1% penicillin/streptomycin (5 mL of 100x penicillin/streptomycin), 12.5% fetal bovine serum (FBS), and 12.5% horse serum. Mix and filter sterilize using a 0.22 µm sterile filtration unit, and store at 4 °C.
- Prepare culture medium for hepatic tumor cell line SK-HEP-1 by adding 10% FBS and 1% penicillin/streptomycin to high-glucose Dulbecco's modified Eagle medium (DMEM) containing 2 mM L-glutamine.
2. Colorimetric LDH Measurement-based NK Cell-mediated Cytotoxicity Assay
- Grow SK-HEP-1 cells to 70−80% confluency in a 100 mm cell culture Petri dish in 5% CO2 at 37 °C in a CO2 incubator. Generate a single-cell suspension by first washing cells with 5 mL of 1x phosphate-buffered saline (PBS) followed by incubation with 1 mL of 0.25% trypsin-EDTA in 5% CO2 at 37 °C in a CO2 incubator until a single cell suspension is generated.
NOTE: NK cell activity can be induced by stressed cancer cells due to changes in expression of NK cell-activating ligand in cancer cells. Therefore, healthy and sub-confluent cancer cells should be used for accurate results. It is also important to note that NK cell receptors and ligands can be sensitive to trypsinization26,27. Therefore, the trypsinization protocol should be carefully optimized and excessive trypsinization should be avoided. - After trypsinization, add 10 mL of SK-HEP-1 culture medium and centrifuge at 160 x g for 3 min in a 15 mL sterile conical centrifuge tube. Wash the cell pellet with 5 mL of 1x PBS and resuspend in 5 mL of culture medium.
- In parallel, centrifuge the NK92MI cells at 160 x g for 3 min in a 15 mL sterile centrifuge tube. Wash the cell pellet with 5 mL of 1x PBS and resuspend in 5 mL of NK92MI cell culture medium.
NOTE: NK92MI cells are grown in NK cell culture medium and obtained similarly as described in step 2.1. - Count the SK-HEP-1 and NK92MI cells using a hemocytometer or any available automated cell counter.
- Add SK-HEP-1 cells (target cells) (1 x 104/100 µL/well) and NK92MI cells (effector cells) (1 x 105/100 µL/well) in the ratio of 1:10 target:effector and seed in triplicate wells in a 96 well plate.
- Incubate the 96 well plate at 37 °C in an atmosphere of 95% air and 5% CO2 for 3 h. After incubation, centrifuge plate at 450 x g for 5 min at room temperature.
- Without disturbing the cell pellet, collect 100 µL of supernatant from each well and transfer to a well in a new 96 well plate.
- Add 50 µL of LDH substrate, mix well, and incubate the plate for 20 min at room temperature in the dark.
- Stop the reaction by adding 50 µL of stop solution (50% dimethylformamide and 20% sodium dodecyl sulfate at pH 4.7). Immediately measure the absorbance of the plate at 490 nm and 680 nm using a plate reader.
- Subtract the absorbance at 680 nm from the absorbance at 490 nm. Calculate the percent (%) NK cell cytotoxicity using the formula below.
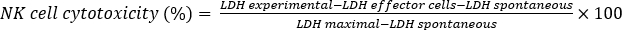
where LDH experimental (effector + target cells) is the absorbance of NK92MI cells and SK-HEP-1 cells, LDH effector cells is the absorbance of NK92MI cells alone, LDH spontaneous is the absorbance of SK-HEP-1 cells alone, and LDH maximal is the absorbance of SK-HEP-1 cells with lysis buffer.
NOTE: To reduce serum interference, always use the following controls: target cells alone (SK-HEP-1), effector cells alone (NK92MI), target cells with lysis buffer as a complete lysis control, target cell medium, NK92MI medium, as well as target cell medium and NK92MI medium in a 1:1 ratio.
3. Calcein AM Staining-based Microscopic Method for Measuring NK Cell-mediated Cytotoxicity
- Culture SK-HEP-1 cells to 70−80% confluency. Generate a single-cell suspension by first washing cells with 5 mL of 1x PBS followed by incubation with 1 mL of 0.25% trypsin-EDTA.
- Centrifuge SK-HEP-1 cells in a 1 mL sterile centrifuge tube at 160 x g for 3 min. Resuspend the pellet in 3 mL of serum-free DMEM.
- Add 1.5 µL of calcein AM solution (10 mM) to SK-HEP-1 cells and incubate for 30 min at room temperature. Centrifuge calcein AM-labeled SK-HEP-1 cells at 160 x g for 3 min in a 15 mL sterile centrifuge tube.
- Wash cells twice with 5 mL of 1x PBS to remove excess calcein AM dye.
- In parallel, centrifuge NK92MI cells at 160 x g for 3 min in a 15 mL sterile centrifuge tube. Wash the cell pellet once with 5 mL of 1x PBS and resuspend in 5 mL of NK92MI cell medium.
- Count calcein AM-labeled SK-HEP-1 cells and NK92MI cells using a hemocytometer or an automated cell counter.
- Resuspend SK-HEP-1 cells in culture medium at 1 x 105 cells/mL and NK92MI cells at 1 x 106 cells/mL in NK cell medium.
- Plate calcein AM-labeled SK-HEP-1 cells (target cells) (1 x 104/100 µL/well) with NK92MI cells (effector cells) (1 x 105/100 µL/well) (1:10 target:effector ratio) per well in triplicate wells in a 96 well plate.
- Incubate the 96 well plate at 37 °C in an atmosphere of 95% air and 5% CO2 for 4 h. After incubation, capture fluorescence images of the calcein AM-labeled cells using a fluorescence microscope at 10x magnification. Capture at least 10 different fields of each replicate for each treatment condition.
- Randomly select 10 images for each replicate and count calcein AM-positive labeled target cells incubated with or without NK92MI cells. Calculate % cytotoxicity using the formula below.

NOTE: As controls, use target cells without NK92MI cells and completely lysed target cells as complete lysis control. For complete lysis, incubate the cells in 0.5% Triton X-100 for 1 h (20 µL of 5% Triton X-100 in 200 µL of culture media).
4. NK Cell Migration Assay
- Grow NK92MI cells and centrifuge cells at 160 x g for 3 min in a 15 mL sterile centrifuge tube.
- Wash the cell pellet twice with 5 mL of 1x PBS and resuspend the cells in 3 mL of serum-free NK92MI cell medium. Count NK92MI cells using a hemocytometer or an automated cell counter.
- Plate NK92MI cells (2.5 x 105 cells/100 µL/well) in the upper compartment of transwell permeable chamber (6.5 mm diameter insert and 5 µm pore size).
- In the bottom chamber add 0.6 mL of serum-free medium containing material to be tested for NK cell chemoattractant properties (e.g., conditioned medium, chemokines, cytokines).
NOTE: When preparing conditioned medium, use reduced-serum medium without added serum to eliminate interference from serum proteins in the migration assay. - Incubate the 24 well permeable chambers at 37 °C for 4 h. After 4 h, collect the non-adherent and migrated NK92MI cells from the bottom chamber and transfer them to fluorescence-activated cell sorting (FACS) tubes for further analysis.
NOTE: The time of culture may vary depending on the type of target cells, as well as the amount and kinetics of chemokines produced by the target cells. Therefore, this time should be empirically determined for each cell type, chemokine, and experiment. - Add predetermined number of counting beads for flow cytometry in a volume of 50 µL to each tube containing migrated NK cells. Evaluate the volume of 300 µL/well cell suspension using any flow cytometer capable of automated FACS-based cell counting.
NOTE: Mix or vortex-mix the counting beads for flow cytometry thoroughly each time before use to ensure that a constant number of beads is used to minimize experimental variability. Reverse pipetting is recommended with count beads to maintain accuracy. Use only NK cells and counting beads for flow cytometry as FACS analysis controls. The authors recommend reading at least 10,000 beads + NK cells combined, an amount that has worked well. However, this number may vary depending upon the experimental conditions. Therefore, the combined number of beads + NK cells should be empirically determined for each type of experiment. Additionally, it is important to perform experiments using biological triplicates to achieve statistically significant results and to account for variability between different cell counts. - Calculate absolute number of migrated NK92MI cells using this formula:
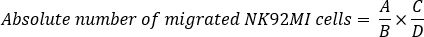
where A = number of cells, B = number of beads, C = assigned bead count of the lot (number of counting beads for flow cytometry/50 µL; in this example 49,500), and D = volume of sample (µL).
NOTE: If 300 µL of sample volume (migrated cells) is used for FACS analysis with 50 µL of counting beads for flow cytometry, the absolute number of migrated cells = 1,700 cells /3,300 bead events x 49,500 beads/300 µL = 84.975 cells/µL. The calculation should be corrected if the sample is diluted or if a different volume of FACS counting beads are used.
NK cell cytotoxicity assays and NK cell migration assay were performed using the SK-HEP-1 hepatic tumor cell line as a model system. To measure NK cell cytotoxicity using the LDH assay, SK-HEP-1 cells expressing either a nonspecific (NS) shRNA or shRNA targeting activating transcription factor 4 (ATF4) were incubated with NK92MI cells in a 96 well plate for 3 h (Figure 1A). ATF4 has been previously shown to regulate NK cell cytotoxicity by upregulating the activating ligand ULBP128. LDH activity associated with NK cell-mediated target cell killing was measured colorimetrically, and percent cytotoxicity calculated using the formula described in protocol 1. ATF4 knockdown significantly reduced NK cell-mediated cytotoxicity compared to NK cells expressing NS shRNA (Figure 1B-D). We also measured NK cell cytotoxicity using a calcein AM staining-based assay. To this end, SK-HEP-1 cells expressing NS shRNA or ATF4-targeting shRNAs were labelled with calcein AM and incubated with NK92MI cells in 96 well plates for 4 h as illustrated in Figure 2A. After incubation, images of calcein AM-positive cells were captured by fluorescence microscopy using a FITC/GFP filter. Cells killed by NK cells are not detected by this approach because they no longer retain the calcein AM dye. As shown in Figure 2B,C, the number of calcein AM-positive SK-HEP-1 cells is decreased after co-culture with NK92MI cells compared to SK-HEP-1 cells grown without NK92MI cells. However, as expected, ATF4 knockdown via shRNA reduced NK cell-mediated killing of SK-HEP-1 cells, observed by a greater number of calcein AM-positive cells (Figure 2B,C). Therefore, both LDH-based and calcein AM assays showed consistent results and confirmed that ATF4 knockdown reduces NK-mediated cancer cell cytotoxicity. Either of these assays is sufficient to assess NK cell-mediated cytotoxicity; however, we recommend using both methods to increase both the stringency and confidence in the results.
We also present the results of a 24 well NK cell migration assay. NK92MI cells were resuspended in serum-free NK92MI medium in the upper chamber, and chemokine, CC motif, ligand 2 (CCL2) was added to the lower permeable chamber. NK cell migration was assayed as described in section 3 (Figure 3A). The number of migrated NK92MI cells was quantified by adding counting beads for flow cytometry and followed by FACS analysis. As shown in Figure 3B-C, there was a significant increase in the number of NK92MI cells that had migrated toward CCL2-containing medium compared to control medium.
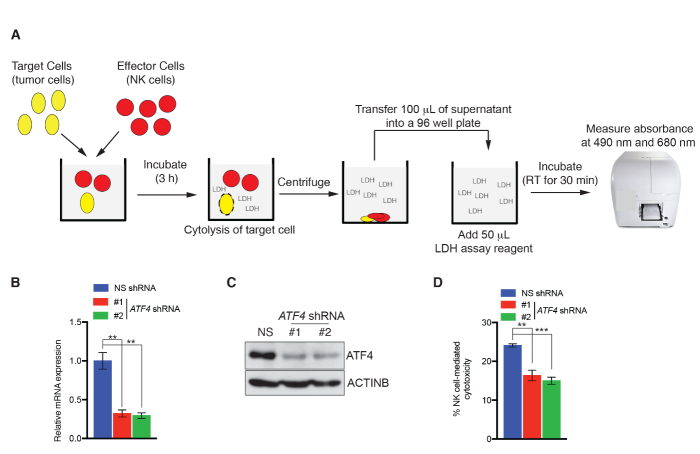
Figure 1: Colorimetric LDH activity-based NK cell-mediated cytotoxicity assay. (A) Schematic depicting the key steps of colorimetric LDH activity-based NK cell cytotoxicity assay. (B,C) ATF4 expression was analyzed in SK-HEP-1 cells expressing either nonspecific (NS) shRNA or shRNAs targeting ATF4 by quantitative RT-PCR and western blotting. (B) Relative ATF4 mRNA level is shown after normalization to ACTINB level in SK-HEP-1 cells expressing NS shRNA or ATF4 shRNAs. (C) ATF4 and ACTINB protein levels in SK-HEP-1 cells expressing NS shRNA or ATF4 shRNAs. (D) NK cell-mediated cytotoxicity was analyzed in SK-HEP-1 cells expressing either NS shRNA or ATF4 shRNAs by the LDH method. Percent (%) NK cell-mediated cytotoxicity is shown. Data are presented as mean ± SEM; ns, not significant; **p < 0.01, ***p < 0.001. Please click here to view a larger version of this figure.
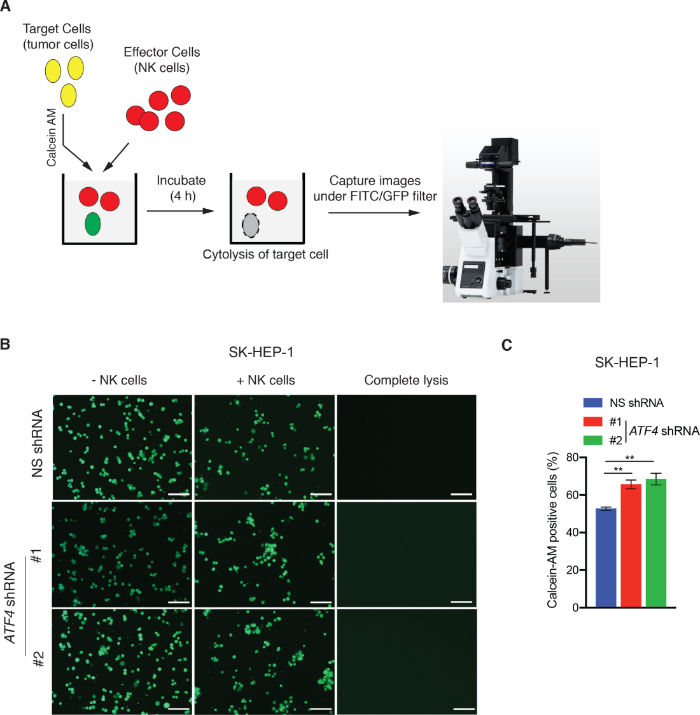
Figure 2: Calcein AM staining-based microscopic method for measuring NK cell-mediated cytotoxicity. (A) Schematic depicting the key steps of calcein AM staining-based microscopic method for measuring NK cell-mediated cytotoxicity. (B) NK cell-mediated cytotoxicity was analyzed in SK-HEP-1 cells expressing either NS shRNA or ATF4 shRNAs using the calcein AM method. Representative images are shown. Scale bar = 200 μm. (C) Percent of calcein AM-positive cells for the experiment presented in panel B. **p < 0.01. Please click here to view a larger version of this figure.
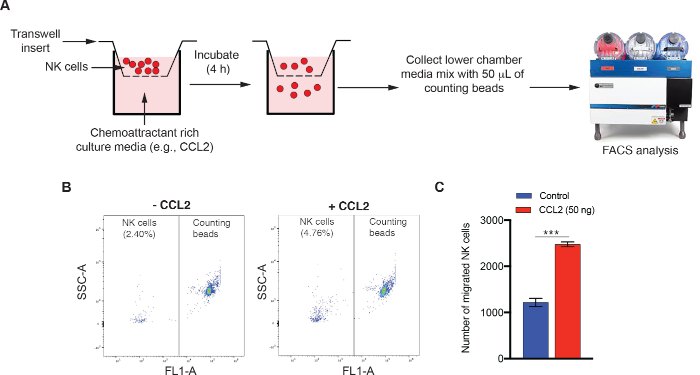
Figure 3: FACS-based quantitative NK cell migration assay. (A) Schematic depicting the key steps of FACS-based NK cell migration assay. (B,C) NK cell migration assay was carried out after adding 50 ng of CCL2 to the bottom chamber of the 24 well plate. (B) Representative NK cell migration dot plots for control or CCL2-treated culture medium are shown. (C) NK cell migration data (mean ± SEM) is presented for the experiment shown in panel B. ***p < 0.001. Please click here to view a larger version of this figure.
