A Neurite Outgrowth Assay and Neurotoxicity Assessment with Human Neural Progenitor Cell-Derived Neurons
Summary
The presented protocol describes a method for a neurite outgrowth assay and neurotoxicity assessment of small molecule compounds.
Abstract
Neurite outgrowth assay and neurotoxicity assessment are two major studies that can be performed using the presented method herein. This protocol provides reliable analysis of neuronal morphology together with quantitative measurements of modifications on neurite length and synaptic protein localization and abundance upon treatment with small molecule compounds. In addition to the application of the presented method in neurite outgrowth studies, neurotoxicity assessment can be performed to assess, distinguish and rank commercial chemical compounds based on their potential developmental neurotoxicity effect.
Even though cell lines are nowadays widely used in compound screening assays in neuroscience, they often differ genetically and phenotypically from their tissue origin. Primary cells, on the other hand, maintain important markers and functions observed in vivo. Therefore, due to the translation potential and physiological relevance that these cells could offer neurite outgrowth assay and neurotoxicity assessment can considerably benefit from using human neural progenitor cells (hNPCs) as the primary human cell model.
The presented method herein can be utilized to screen for the ability of compounds to induce neurite outgrowth and neurotoxicity by taking advantage of the human neural progenitor cell-derived neurons, a cell model closely representing human biology."
Introduction
Neurite growth is a process fundamental to the formation of the neuronal network and nerve regeneration1,2. Following an injury, neurite outgrowth plays a key role in regeneration of the nervous system. Neurite outgrowth is also an important element of the extracellular signaling in inducing neuronal regenerative activities to enhance the outcomes for neurodegenerative disorders and neuronal injury3,4,5,6.
By maintaining their differentiation potential in producing various neural lineages, human neural progenitor cells (hNPCs) could provide a model system for studies of central nervous system (CNS) function and development7,8,9. High translational potential and physiological relevance of hNPCs as a primary human cell model offer a considerable advantage in neurite outgrowth-related drug discovery screenings. However, the maintenance and scaling of the primary cell models for high-throughput assays could be time-consuming and labor-intensive10,11,12,13.
In addition to the application of the presented method in neurite outgrowth studies, neurotoxicity assessment is another application using the hNPC-derived neurons. There are thousands of commercial chemical compounds that are either not examined or with poorly understood neurotoxicity potential. Therefore, more reliable and effective screening experiments to assess, distinguish, and rank compounds based on their potential to elicit developmental neurotoxicity is in high demand14. The increase in prevalence and incidence of neurological disorders along with the abundance of untested compounds in the environment necessitates the development of more trustworthy and efficient experiments to identify hazardous environmental compounds that may pose neurotoxicity15.
The presented method herein can be utilized to screen for the ability of compounds to induce neurite outgrowth and neurotoxicity by taking advantage of the human neural progenitor cell-derived neurons, a cell model closely representing human biology.
Protocol
Ethics Statement: Fetal specimens were received from the Birth Defects Research Laboratory at the University of Washington in Seattle through a tissue distribution program supported by the National Institute of Health (NIH). The Birth Defects Research Laboratory obtained appropriate written informed consent from the parents and the procurement of tissues was monitored by the Institutional Review Board of the University of Washington. All the work was performed with approval by the Human Subject Research Office at the University of Miami8.
1. Isolation and culture of human neural progenitor cells (hNPCs)
- Place the brain tissue in a 100 mm Petri dish and carefully remove the meninges using forceps.
- Transfer the brain tissue to a 50 mL conical tube and wash it twice with 20 mL of PBS by gently inverting the tube.
- Incubate the brain tissue in a new 50 mL conical tube by submerging the tissue in cell dissociation solution (see Table of Materials) and DNase I (10 U/mL) for 10 min at 37 °C.
- Add 5 mL of neuronal cell culture medium (see Table of Materials) to the brain tissue containing tube and mechanically dissociate the neurospheres by triturating 20 to 30 times through a 1000 µL pipet tip to make a single-cell suspension.
- Filter the cell suspension through a 70 µm cell strainer to remove cell clusters.
- Seed the single-cell suspension in a vented T-25 flask provided with 5-10 mL of neuronal cell culture medium supplemented with components detailed in Table 1.
- Sterilize the heparin solution by filtration through a 0.2 µm filter. The B-27 supplement without Vitamin A is a serum-free supplement for the cultivation of neural progenitors and stem cells, without inducing differentiation.
NOTE: Human neural progenitor cells (hNPCs) are isolated from human fetal brain collected from aborted fetus. Following 7–10 days in culture, neural stem cells (NSCs) form free-floating neurosphere colonies, whereas other cell types remain in suspension as single cells or attach to the bottom of the flask. The isolated hNPCs can be cultured as neurospheres in suspension for several months8,16,17.
- Sterilize the heparin solution by filtration through a 0.2 µm filter. The B-27 supplement without Vitamin A is a serum-free supplement for the cultivation of neural progenitors and stem cells, without inducing differentiation.
| Amount | Component |
| 100 µL | EGF (20 ng/mL) |
| 100 µL | FGF (10 ng/mL) |
| 2 mL | B-27, Minus vitamin A (50X) |
| 1 mL | L-alanyl-L-glutamine (100X) (see table of materials) |
| 4 µL | Heparin (2 μg/mL) |
| 96.8 mL | Neuronal cell culture medium (see table of materials) |
Table 1. Components required for making 100 mL of culture media
2. Passaging the hNPCs
- Collect the media containing the floating spheres, big and small neurospheres, and transfer them to a 50 mL conical tube.
NOTE: Due to unknown reasons, the timing for splitting the neurospheres is variable from 7 days up to 30 days. However, generally, neurospheres need to be passaged when they reach a diameter greater than 700-900 µm. This is when the center of the neurosphere starts to darken, which is considered as a sign of a high rate of cell death18. - Spin the neurospheres down by centrifugation at 300-400 x g for 3 min.
- Carefully aspirate the supernatant and then submerge the spheres in 500 µL of defrosted cell dissociation reagent (see Table of Materials).
- Make aliquots of cell dissociation reagent by adding 500 µL per 1.5 mL microtubes and store at -20 °C. To avoid losing enzyme activity, thaw the cell dissociation reagent by holding at RT or warm for 5 min in a 37 °C water/bead bath.
- Depending upon the density and size of the spheres, incubate the submerged spheres at 37 °C for 5-15 min.
- Add 5-10 mL of pre-warmed culture media to the neurospheres containing 50 mL conical tube and centrifuge at 300-400 x g for 5 min to sediment the neurospheres.
- Aspirate the supernatant and gently pipette up and down, using a 1000 µL pipette, in 2 mL of culture media until all the neurospheres are in a single cell suspension.
NOTE: The dissociation will become visible to the naked eye. Before dissociation, the neurospheres are in the form of spheres. After submerging in dissociation reagent and by pipetting up and down, they will become single cells.- Count the cells and plate the single cells, 2 to 3 million cells per T-25 flask in 10 mL of culture media.
- Feed the cells every 3 days by replacing half the culture media.
- Settle neurospheres by leaning the flask so that it is on its bottom corner. Hold the flask in the position for about 1-2 min until the neurospheres sediment. Then aspirate half the media gently by inserting the serological pipette in the media above the settled neurospheres. Dissociated cells can aggregate to form spheres after 2 to 3 days in culture19.
3. Freezing the hNPCs
- Prepare the cell freezing medium by adding DMSO to the culture media to a final concentration of 10% (v/v) or use commercially available cryopreservation medium for sensitive cell types (see Table of Materials).
- Sort out big spheres by transferring the media into a 50 mL conical tube and letting the spheres settle by gravity. Then remove and transfer the big neurospheres into a new 50 mL conical tube for passaging using a 200 or 1000 µL pipet tip.
NOTE: Neurospheres with a diameter greater than 900 µm are considered big and the ones with a diameter smaller than 500 µm are considered small. - Spin the remaining neurospheres down by centrifugation at 300-400 x g for 3 min. Carefully remove the supernatant.
- Resuspend up to 100 spheres in 1 mL of cryopreservation reagent and transfer it to a cryotube.
- Store overnight at -80 °C in a cell freezing container (see Table of Materials) and move it to liquid nitrogen the next day for long-term storage.
NOTE: It is preferable to freeze small to medium-sized neurospheres (lower than 900 µm in diameter) and avoid freezing big-sized neurospheres (greater than 900 µm in diameter) or single cells. In order to reduce cell damage during thawing the sample, keep the cells dense by seeding the thawed neurospheres into a small T25 flask.
4. Differentiation and treatment of hNPCs
NOTE: To induce differentiation, neurospheres are disaggregated into single cells, counted and then seeded on coated plates for 5 days. Then differentiated cells are treated for 24 h with test compounds before immunostaining and fluorescence quantification.
- Coating
- Add 200 µL of poly-L-lysine (PLL) per well of 4-well glass chamber slides (140 µL per well of 8-well chamber slides).
- Incubate for 1 h at room temperature (RT).
- Wash 3x with PBS.
- Let it dry at RT (for about 30 min).
- Add 150 µL of laminin (50 µg/mL) per well of 4-well glass chamber slides (120 µL per well of 8-well chamber slides).
- Incubate for 2 h at 37 °C.
- Wash 3x with PBS.
NOTE: Coated chamber slides can be stored at 4 °C for 1 month.
- Plating the cells
- Count and plate 80,000 single cell neurospheres (neurospheres in a single cell suspension) per well of 4-well chamber slides (70,000 cells per well of 8-well chamber slide).
- Add 500 µL of differentiation media per well of 4-well chamber slide (250 µL media per well of 8-well chamber slides).
- To make the differentiation media first, add the following components in Table 2 to a sterile, disposable container to make the neural inducing media (NIM).
- Add the following ingredients in Table 3 to 48.5 mL of NIM made in the previous step to make the differentiation media.
- Incubate for 5 days at 37 °C.
- After 5 days, treat the cells for 24 h by replacing half the media in each well with fresh media mixed with the desired concentration of test compounds, including appropriate controls.
| Amount | Component |
| 49 mL | DMEM/F-12 |
| 0.5 mL | N2 supplement (100X) |
| 0.5 mL | MEM non-essential amino acids (100X) |
| 2 µL | Heparin (2 µg/mL) (Stock Conc. is 50 mg/mL) |
Table 2. Components required for making 50 mL of NIM
| Amount | Component |
| 1 mL | B-27 (50X) |
| 500 µL | Antibiotic-Antimycotic (100X) |
| 5 µL | Retinoic acid (0.1 µM) |
| 50 µL | GDNF (10 µg/mL) |
| 50 µL | BDNF (10 µg/mL) |
| 5 µL | Ascorbic acid (0.2 µg/mL) (Stock Conc. is 2 mg/mL) NOTE: Recommended to be made fresh. |
| 48.5 mL | NIM |
Table 3. Components required for making 50 mL of differentiation media
5. Immunocytochemistry (ICC)
NOTE: Cells are fixed with 4% formaldehyde. Permeabilization and blocking is then performed to improve penetration and prevent nonspecific binding of antibodies. Cells are then incubated with primary antibodies overnight. Subsequently, cells are incubated with fluorescently labeled secondary antibodies. Finally, after using DAPI to stain the nucleus, chamber slides are mounted.
- Fixation
- Gently aspirate the media in each well.
- Add 500 µL of 4% formaldehyde per well of 4-well chamber slides (250 µL per well of 8-well chamber slides).
- Incubate for 15 min at RT.
- Gently wash 2x with 500 µL of PBS.
NOTE: After fixation, by leaving 1 mL of PBS in each well, culture slides can be stored at 4 °C for up to 3 months.
- Cell permeabilization and blocking
- Add the following components in Table 4 to a sterile, disposable container to make the antibody (Ab) buffer.
- Mix the following ingredients in Table 5 with the Ab buffer made in the previous step to make the cell permeabilization and blocking solution.
- Add 500 µL per well of 4-well chamber slides and incubate for 1 h at RT.
NOTE: Ab buffer can be stored at 4 °C.
| Amount | Component |
| 1.75 g | NaCl (150 mM) |
| 1.2 g | TrisBase (50 mM) |
| 2 g | BSA 1% |
| 3.6 g | L-lysine (100 mM) |
| 8 g | Sodium Azide (4%) |
| 200 mL | Distilled water. NOTE: Initially dissolve the required components in 150 mL of water, then adjust to 200 mL. |
Table 4. Components required for making 200 mL of Antibody buffer
| Amount | Component |
| 600 µL | 20% Goat serum |
| 6 µL | 0.2% Triton-X100 |
| 2394 µL | Antibody buffer. NOTE: Initially dissolve the required components in 2 mL of Ab buffer, and then adjust to pH 7.4. Then add more Ab buffer to adjust to final volume of 3 mL and filter sterilize. |
Table 5. Components required for making 3 mL of cell permeabilization and blocking solution
- Staining
- Wash 2x with 500 µL of PBS.
- Add the diluted primary antibody, anti-β-tubulin III (1:200), and incubate overnight at 4 °C (200 µL per well of 4-well chamber slides). Dilute the primary antibody in PBS.
- Wash 2x with PBS.
- Add the diluted, in PBS, fluorescently labeled secondary antibody, Alexa Fluor 488 (1:500), and incubate for 2 h at RT in a place protected from light (250 µL per well of 4-well chamber slides)
- Wash 2x with PBS.
- Add the diluted, in PBS, DAPI (300 nM concentration) and incubate for 5 min at RT in a place protected from light (300 µL per well of 4-well chamber slides).
- Wash 3x with PBS.
- Mount the chamber slides using the following instructions.
- Take apart the chamber slide by breaking the breakaway tabs and removing the gasket and base.
- Add one drop of the mounting solution (see Table of Materials) per well of 4-well chamber slides. Then use a cover slide to cover the whole slide.
- Using a tweezer and at an angle, place one side of the cover slip against the slide while making contact with the outer edge of the liquid drop.
- Carefully tip the coverslip onto the mounting solution when lowering it into place. Avoid the creation of bubbles. Take a pipette tip and press it down on the cover slip. The bubbles will move to the side.
NOTE: Bubble formation is inevitable at times. If it occurs, image around them as long as there are a few. - Follow manufacturer’s directions for curing time. Avoid using noncuring mounting solutions due to the difficulty in handling during imaging. Otherwise using an appropriate coverslip sealant on edges is required to prevent the coverslip from sliding during imaging.
- To seal the coverslip, use nail polish and make a small line on the edge of the cover slip. Let the nail polish dry for about 2 min.
6. Image acquisition, neurite outgrowth and fluorescence intensity quantification
NOTE: Following staining, use a confocal microscope with a 20x objective and an image size of 1024 x 1024 pixels to acquire the images of the treated cells. Take image at least from two fields per biological replicate per condition. Then use Fiji image analysis software (ImageJ 1.51u) for quantification of the neurite length. Briefly, measure the length of the longest neurite for each neuron and after averaging the values per treatment, use student’s t test for independent groups to compare the means between experimental groups and control group.
NOTE: Several commercial (Imaris, Volocity, Amira) and open source (ImajeJ, CellProfiler, Vaa3D, BioImageXD, Icy, KNIME) image processing programs are available. Among these programs, ImageJ has become the tool of choice for biological image analysis20,21. The ImageJ portal at https://imagej.net/Introduction is a useful source of information providing a thorough description of ImageJ’s basic, and built-in functions including image processing, colocalization, deconvolution, registration, segmentation, tracking, and visualization.
- Measuring neuronal outgrowth with Fiji image analysis software
- As illustrated in Figure 1, open the image either by dragging and dropping it onto Fiji software or by selecting File | Open.
- Select Analyze | Tools | ROI manager, and then right-click on the 5th icon in the toolbar Straight and switch to freehand line. Optionally, double-click on the same icon to change the line width to 10, and then trace the longest neurite, beginning near the cell body and extending to the tip of the neurite.
- Press Ctrl + T & then F to add the measurement to the ROI Manager and highlight the measured neuron. Select all numbers in the ROI, click on Measure, select all calculated lengths, and copy/paste into a spreadsheet.
- Measuring fluorescence intensity of labeled cells with Fiji image analysis software
- As illustrated in Figure 2, after opening the image, click on the 4th icon in the toolbar Freehand selections. Draw the shape of the cell.
- Select Analyze | Set Measurements and select for the following values: Area, Integrated density, Mean grey value | Analyze | Measure (a pop-up box with a stack of values opens).
- Select a region next to the cell as background (size is not important) and then select Analyze | Measure. Select all measured data and copy/paste into a spreadsheet.
NOTE: Integrated Density (IntDen) is the item used for determining the fluorescent intensities. In this study fluorescent intensity of the target molecule is measured to initially analyze its distribution in the cell and subsequently quantifying the abundance of the target molecule under various treatments. Consequently, the effectiveness of treatments will be measured and compared with control.
7. Neurotoxicity assessment
NOTE: Cytotoxicity of test compounds are evaluated in 384-well plates (see Table of Materials) using a luminescent cell viability assay (see Table of Materials). The hNPCs are prepared following the same method, except slight modifications, described in the “Differentiation and Treatment of hNPCs” section. Subsequently, a luminescent signal generated by luminescent cell viability assay is measured utilizing a microplate reader. The luminescent signal is proportional to the cellular ATP concentration which itself is directly proportional to the number of viable cells present in each well.
- Coating
- Add 30 µL of poly-L-lysine (PLL) per well of 384-well plate.
- Incubate for 1 h at RT.
- Wash 2x with PBS.
- Let it dry at RT (for about 30 min).
- Add 30 µL of laminin (50 µg/mL) per well of 384-well plate.
- Incubate for 2 h at 37 °C.
- Wash 2x with PBS.
NOTE: Coated 384-well plates can be stored at 4 °C for 1 month.
- Plating the cells
- Count and plate 20,000 single cell neurospheres per well of 384-well plate in 25 µL of differentiation media.
- Incubate for 5 days at 37 °C.
- After 5 days, treat the cells for 24 h by test compounds prepared at 6x the final desired concentration in 5 µL volume (to make the final volume of 30 µL per well).
- Cell viability assay
- Add 30 µL of luminescent cell viability assay reagent per well of 384-well plate.
NOTE: Add a volume of luminescent cell viability assay reagent equal to the volume present in each well. Thaw the luminescent cell viability assay reagent and equilibrate to RT prior to use. - Shake on a plate shaker for 2 minutes (to mix and induce cell lysis).
- Spin the mixture down by centrifugation at 300-400 x g for 30 s.
- Incubate the 384-well plate for 10 min at RT in a place protected from light (to stabilize the luminescent signal).
- Record luminescence with a microplate reader.
NOTE: Use appropriate controls for the viability assay including Velcade (at a final concentration of 10 µM) as positive and HBSS containing DMSO (with final concentration of 0.1% or 0.2%) as negative control.
- Add 30 µL of luminescent cell viability assay reagent per well of 384-well plate.
Representative Results
The protocol presented in the manuscript has successfully been used in two recently published papers22,23. Figure 3 demonstrates the use of hNPCs-derived neurons in examining the effect of HDAC inhibitors as epigenetic compounds on the extension of neurites as a marker for neurite outgrowth and subsequent neurogenic ability of small molecule compounds.
Furthermore, in Figure 4 the neurotoxicity of tested compounds (HDAC inhibitors) is also assessed by simultaneously differentiating the hNPCs in 384-well plates, showing the potential of the presented cell model (hNPCs) for neurotoxicity assessment and the ability to scale up to test neurotoxicity for a higher number of compounds.
In another paper by Sartor et al. (Figure 5), measurement of the neuronal cell fluorescence intensity to quantify the abundance of H4K5ac, as a histone mark, after treating the neurons differentiated from hNPCs with epigenetic modifier compounds is successfully demonstrated23. Additionally, as illustrated in Figure 6, in an unpublished work, the protocol has been used to visualize a presynaptic protein, synaptophysin, to check for the possible synaptogenic effect of small molecule compounds.
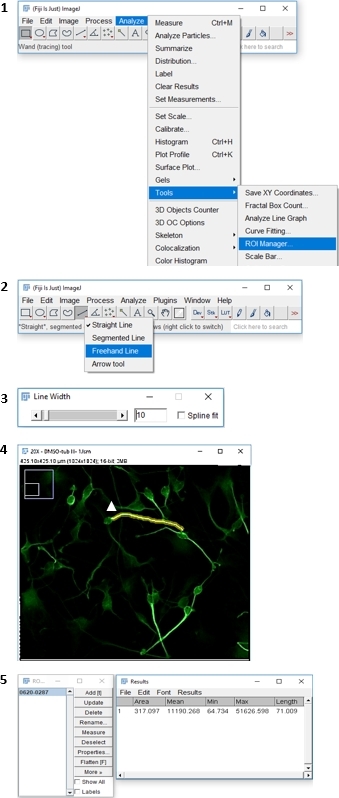
Figure 1: Step-by-step workflow illustrating an approach for measuring neuronal outgrowth with Fiji image analysis software. Please click here to view a larger version of this figure.
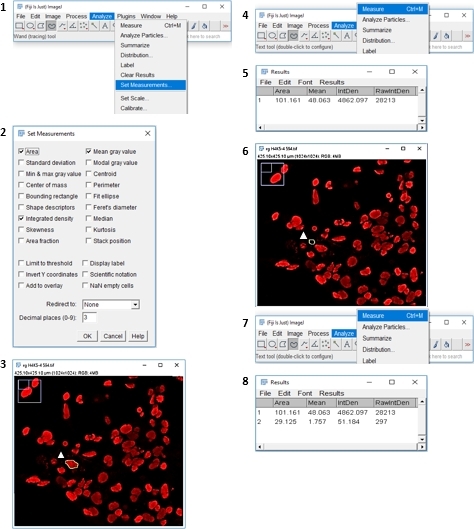
Figure 2: Step-by-step workflow illustrating an approach for quantifying neuronal cells fluorescence intensity with Fiji image analysis software. Please click here to view a larger version of this figure.
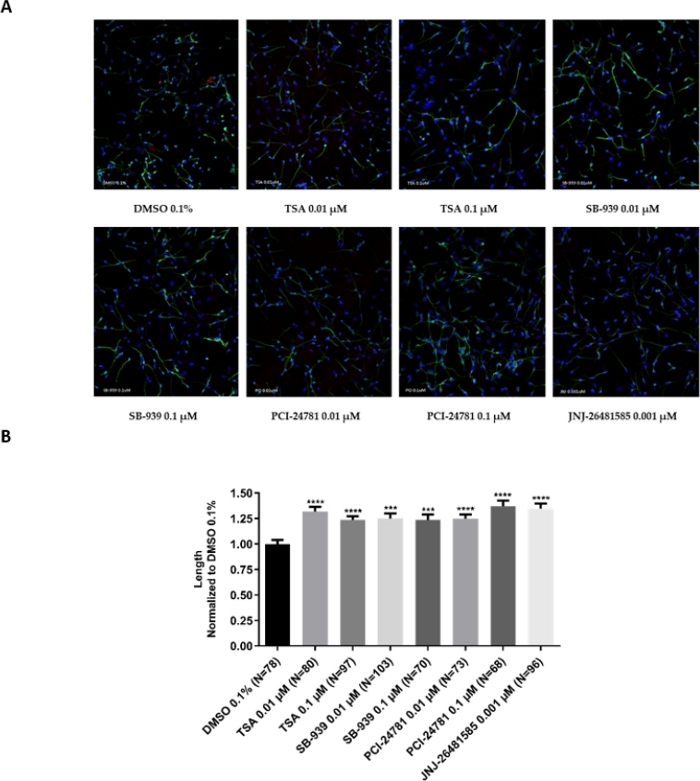
Figure 3: Neurite outgrowth assay with small molecule epigenetic compounds.
(A) Representative fluorescent images (20x magnification) for treatment of hNPCs-derived neurons with hydroxamic-based HDAC inhibitors. Human neural progenitor cells are differentiated for 5 days; then treated for 24 h with 0.1% DMSO (as control), and Trichostatin A, JNJ26481585, SB939, and PCI24781 (as test compounds). Then immunostaining is performed with neuronal-specific β-tubulin III antibody (green) to visualize neuronal processes and quantify neurite lengths and DAPI (blue) to visualize cell nuclei. (B) Statistical analysis of neurite length in various groups. As depicted, all test compounds can induce neurite outgrowth. Quantitative analysis of neurite length is done using ImageJ software. Error bars represent SEM; *** p < 0.001, **** p < 0.0001. This figure has been modified from Bagheri et al.22. Please click here to view a larger version of this figure.
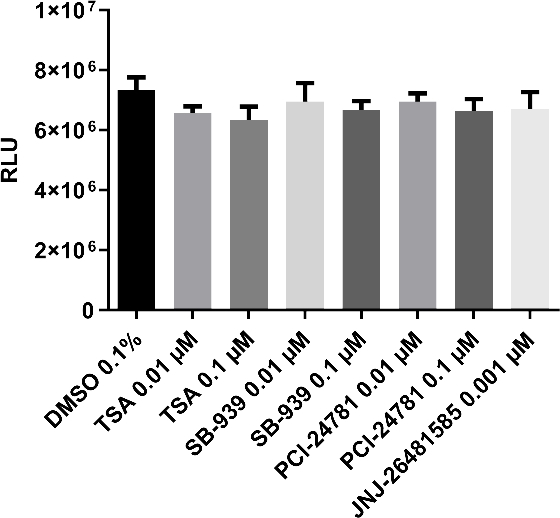
Figure 4: Neurotoxicity assessment using luminescent cell viability assay.
hNPCs are seeded and differentiated to neurons in the 384-well plate according to the same method used for neurite outgrowth assay. Thereafter the effect of test compounds on the viability of cells after 24 h of exposure is measured. As shown, there is no significant toxicity upon treatments. One-way ANOVA is used for data analysis and a p value < 0.05 is considered statistically significant. This figure has been modified from Bagheri et al.22. Please click here to view a larger version of this figure.
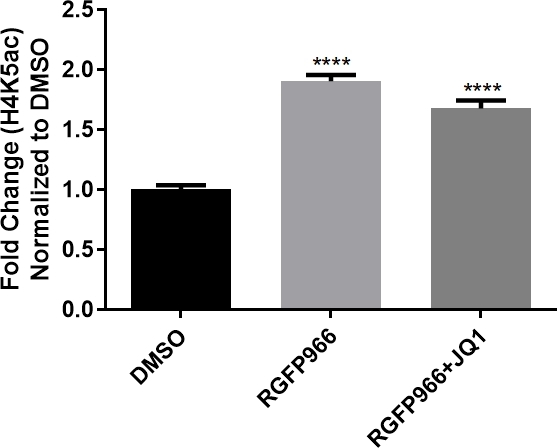
Figure 5: RGFP966 increases H4K5ac in neurons differentiated from human neural progenitor cells.
(A) Representative immunofluorescence staining of β tubulin, DAPI, and H4K5ac following treatment with DMSO, RGFP966, or RGFP966 + JQ1 in hNPCs-derived neurons. (B) Quantification of fluorescence intensity of H4K5ac following treatment with DMSO, RGFP966, or RGFP966 + JQ1. Error bars represent SEM; **** p < 0.0001. This figure has been modified from Sartor et al.23. Please click here to view a larger version of this figure.
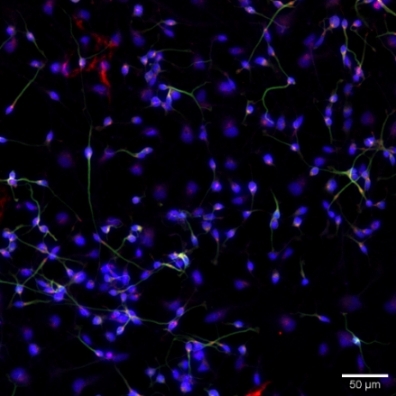
Figure 6: Representative immunofluorescence staining of Synaptophysin (red) to visualize synaptic terminals, β tubulin (green) to visualize neuronal processes, and DAPI (blue) to visualize cell nuclei following treatment with DMSO (as control), and SB939 (as test compound) in hNPCs-derived neurons. Please click here to view a larger version of this figure.
Discussion
This protocol is one of the few published papers describing the test for neurite length upon treatment with test compounds. Furthermore, we describe how to use hNPCs for a neurite outgrowth assay and neurotoxicity assessment. By utilizing this neurite outgrowth assay and neurotoxicity assessment on hNPCs-derived neurons, the neurogenic potential of a category of epigenetic small-molecule compounds, HDAC inhibitors, in inducing neurite outgrowth is demonstrated22. Furthermore, in another paper presented by Sartor et al., this protocol is used to quantify the fluorescence intensity of H4K5ac, a histone protein, upon treatment with several epigenetic modifier compounds23.
Cognitive function relies on proper wiring and functional connections within neuronal circuits. Detailed and consistent quantification of various aspects of neuronal morphology as a phenotypic screening approach is essential to achieve reliable insight in the underlying pathways leading to brain disorders ranging from neurodevelopmental to psychiatric disorders. Phenotypic screening is considered a notably effective approach in exploring small-molecule compounds as putative drug candidates to modulate a cellular phenotype. In this approach, instead of interrogating only a single target, all components and pathways of the cell are examined24.
Neuronal morphology including shape, structure, and connectivity are considered to be key features of neuronal function. Genetic perturbations that alter the morphology of neurons or synaptic protein localization and level could notably contribute to the analysis of disease-causing mutations. Therefore, reliable methods are required to assess the impact of perturbagens on neuronal morphology25.
The hNPCs, are isolated from the embryonic mammalian brain, which are subsequently cultured in vitro in the presence of mitogens. These cells are made up of neurospheres that are characterized as floating cellular aggregates comprised of neural progenitor cells and radial glial cells. The hNPCs provide a desirable model system for nervous system function and development by maintaining their differentiation potential in producing various neural lineages7,8,9. Neurite number, intensity, length, and width together with synaptic characteristics including pre- and post-synaptic proteins modifications are among neurite and synaptic changes that could be reliably and efficiently measured using the presented method herein.
Due to an increase in the prevalence of neurological disorders such as attention deficit hyperactivity disorder and autism, the neurotoxicity potential of environmental chemicals on children remains a public concern15,26. Therefore, the usability of hNPCs-derived neurons for reliable and efficient neurotoxicity assessment is also examined. As demonstrated in Figure 4, hNPCs could be adapted and scaled up for neurotoxicity assessment in 384-well plates.
Neurotoxicity assessment as an application for the introduced cell model is feasible by either differentiating the neurospheres into neurons before plating in 384-well plates or differentiating the cells while the neurospheres are plated in 384-well plates as single cells. In the latter approach, high precision pipetting skills are required for plating the exact number of neurospheres that are disaggregated into single cells and also for the following treatment procedures with differentiation-inducing medium. Considering the pipetting skills required, differentiation before plating is recommended to achieve more reproducible results. Moreover, bulk cell sorting by fluorescence activated cell sorting (FACS) could also potentially be used to accurately separate the required number of cells circumventing the need for high precision pipetting skills.
It is advised to start the neurite outgrowth assay by neurotoxicity assessment to avoid repeated neurite outgrowth assays with various doses of the same compound to reach for nontoxic concentration. Therefore, combining a neurotoxicity assessment by utilizing the luminescent cell viability assay against a dose-response curve of test compounds will result in finding the right dosage with minimum effort. The luminescent cell viability assay is a highly sensitive method to determine the number of viable cells in a cell culture. The method works on quantitation of the ATP present in culture as an indicator of cellular metabolism. The add, mix, and measure format of the assay provides a simple and rapid approach to check for cell viability and cytotoxicity of test compounds.
There are some limitations to the presented protocol as follows. Due to availability and inherent variability, the use of physiologically relevant primary cells is limited25. hNPCs have a slow proliferation rate, which could limit its utilization. Manual large-scale analysis of fluorescent images of neurons is time-consuming and susceptible to experimenter bias. hNPCs could be considered as young neural cells, not representing age-related epigenetic modifications accumulating in cells throughout a person’s life. As neurodegenerative disorders occur in older individuals, hNPCs may lack adequate physiological relevance to be an appropriate model for late onset conditions.
Even though animal models and cell lines have been valuable in deciphering molecular mechanisms underlying diseases, they have shown limitations in translating findings into human therapeutics27,28,29,30. Human induced pluripotent stem cells (hiPSCs) and human embryonic stem cells (hESCs) alongside hNPCs are considered as better in vitro cell models, recapitulating human physiology. Therefore hiPSCs- and hESCs- derived neurons can also potentially be used as two alternative cell sources for neurite outgrowth assay and neurotoxicity assessment, utilizing the protocol presented herein1,31.
In conclusion, high translational potential and physiological relevance of hNPCs as primary human cell model provides a valuable recourse in neurite outgrowth-related drug discovery screenings and neurotoxicity assessment outweighing the limitations mentioned earlier.
Divulgations
The authors have nothing to disclose.
Acknowledgements
This research was funded by NIMAD research grant (940714) awarded to MAF.
Materials
| 4-well Glass Chamber Slides | Sigma | PEZGS0816 | |
| Alexa Fluor 488 | Invitrogen | A-11001 | |
| Alexa Fluor 594 | Invitrogen | R37117 | |
| Antibiotic-Antimycotic | Gibco | 15240062 | |
| Anti-β-Tubulin III | Thermo | MA1-118X | |
| B27 | Thermo | 17504001 | |
| B27 – minus vitamin A | Thermo | 12587010 | |
| BDNF | PeproTech | 450-02 | |
| BSA | Sigma | A8531 | |
| CellTiter-Glo | Promega | G7572 | |
| CoolCell | Corning | 432000 | Cell freezing containers ensuring standardized controlled-rate -1℃/minute cell freezing in a -80℃ freezer |
| CryoStor CS10 | StemCell Technologies | 7930 | Cryopreservation medium containing 10% DMSO |
| DAPI | Thermo | D1306 | |
| DMEM/F12 | Gibco | 11320033 | |
| DMSO | Sigma | 34869-100ML | |
| EGF | Gibco | PHG0311 | |
| FGF | Gibco | PHG6015 | |
| Formaldehyde | Thermo | FB002 | |
| GDNF | PeproTech | 450-10 | |
| Glutamax | Gibco | 35050061 | L-alanyl-L-glutamine supplement |
| Goat Serum | Thermo | 50062Z | |
| Heparin | Calbiochem | 375095 | |
| Laminin | Sigma | L2020-1MG | |
| L-Ascorbic Acid | Sigma | A92902-25G | |
| L-lysine | Sigma | L5501 | |
| MEM non-essential amino acids | Gibco | 11140050 | |
| mFreSR | StemCell Technologies | 5854 | Serum-free cryopreservation medium designed for the cryopreservation of human embryonic and induced pluripotent stem cells |
| N2 | Gibco | 17502048 | |
| NaCl | Sigma | 71376 | |
| Neurobasal Medium | Gibco | 21103049 | |
| Nunc 384-Well Polystyrene White Microplates | Thermo | 164610 | |
| PBS | Thermo | 10010-049 | |
| Poly‐L‐lysine | Sigma | P5899-5MG | |
| ProLong Gold Antifade Mountant | Thermo | P10144 | |
| Retinoic Acid | Sigma | R2625 | |
| Sodium Azide | Sigma | S2002 | |
| StemPro Accutase | Gibco | A1110501 | Cell dissociation reagent containing proteolytic and collagenolytic enzymes |
| Synaptophysin | Thermo | MA5-14532 | |
| Tris Base | Sigma | 10708976001 | |
| Triton X-100 | Sigma | X100-100ML |
References
- Sherman, S. P., Bang, A. G. High-throughput screen for compounds that modulate neurite growth of human induced pluripotent stem cell-derived neurons. Disease Models & Mechanisms. 11 (2), (2018).
- Al-Ali, H., Beckerman, S. R., Bixby, J. L., Lemmon, V. P. In vitro models of axon regeneration. Experimental Neurology. 287, 423-434 (2017).
- Kudo, T., et al. Induction of neurite outgrowth in PC12 cells treated with temperature-controlled repeated thermal stimulation. PloS One. 10 (4), 0124024 (2015).
- Higgins, S., Lee, J. S., Ha, L., Lim, J. Y. Inducing neurite outgrowth by mechanical cell stretch. BioResearch Open Access. 2 (3), 212-216 (2013).
- Muramatsu, R., Ueno, M., Yamashita, T. Intrinsic regenerative mechanisms of central nervous system neurons. Bioscience Trends. 3 (5), (2009).
- Read, D. E., Herbert, K. R., Gorman, A. M. Heat shock enhances NGF-induced neurite elongation which is not mediated by Hsp25 in PC12 cells. Brain Research. 1221, 14-23 (2008).
- Finan, G. M., et al. Bioactive Compound Screen for Pharmacological Enhancers of Apolipoprotein E in Primary Human Astrocytes. Cell Chemical Biology. 23 (12), 1526-1538 (2016).
- Magistri, M., et al. A comparative transcriptomic analysis of astrocytes differentiation from human neural progenitor cells. European Journal of Neuroscience. 44 (10), 2858-2870 (2016).
- Bez, A., et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Research. 993 (1-2), 18-29 (2003).
- Grandjean, P., Landrigan, P. J. Neurobehavioural effects of developmental toxicity. The Lancet Neurology. 13 (3), 330-338 (2014).
- Finkbeiner, S., Frumkin, M., Kassner, P. D. Cell-based screening: extracting meaning from complex data. Neuron. 86 (1), 160-174 (2015).
- An, W. F., Tolliday, N. Cell-based assays for high-throughput screening. Molecular Biotechnology. 45 (2), 180-186 (2010).
- Astashkina, A., Mann, B., Grainger, D. W. A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacology & Therapeutics. 134 (1), 82-106 (2012).
- Swinney, D. C., Anthony, J. How were new medicines discovered. Nature Reviews Drug Discovery. 10 (7), 507 (2011).
- Ryan, K. R., et al. Neurite outgrowth in human induced pluripotent stem cell-derived neurons as a high-throughput screen for developmental neurotoxicity or neurotoxicity. Neurotoxicology. 53, 271-281 (2016).
- Magistri, M., Velmeshev, D., Makhmutova, M., Faghihi, M. A. Transcriptomics profiling of Alzheimer’s disease reveal neurovascular defects, altered amyloid-β homeostasis, and deregulated expression of long noncoding RNAs. Journal of Alzheimer’s Disease. 48 (3), 647-665 (2015).
- Darbinyan, A., Kaminski, R., White, M. K., Darbinian, N., Khalili, K. Isolation and propagation of primary human and rodent embryonic neural progenitor cells and cortical neurons. Neuronal Cell Culture. , 45-54 (2013).
- Gil-Perotín, S., et al. Adult neural stem cells from the subventricular zone: a review of the neurosphere assay. The Anatomical Record. 296 (9), 1435-1452 (2013).
- Ebert, A. D., McMillan, E. L., Svendsen, C. N. Isolating, expanding, and infecting human and rodent fetal neural progenitor cells. Current Protocols in Stem Cell Biology. 6 (1), 2 (2008).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676 (2012).
- Collins, T. J. ImageJ for microscopy. Biotechniques. 43 (1), 25-30 (2007).
- Bagheri, A., et al. HDAC Inhibitors Induce BDNF Expression and Promote Neurite Outgrowth in Human Neural Progenitor Cells-Derived Neurons. International Journal of Molecular Sciences. 20 (5), 1109 (2019).
- Sartor, G. C., et al. Enhancement of BDNF expression and memory by HDAC inhibition requires BET bromodomain reader proteins. Journal of Neuroscience. 39 (4), 612-626 (2019).
- Conde, C., Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nature Reviews Neuroscience. 10 (5), 319 (2009).
- Schmitz, S. K., et al. Automated analysis of neuronal morphology, synapse number and synaptic recruitment. Journal of Neuroscience Methods. 195 (2), 185-193 (2011).
- Grandjean, P., Landrigan, P. J. Developmental neurotoxicity of industrial chemicals. The Lancet. 368 (9553), 2167-2178 (2006).
- Dragunow, M. The adult human brain in preclinical drug development. Nature reviews Drug Discovery. 7 (8), 659 (2008).
- Dolmetsch, R., Geschwind, D. H. The human brain in a dish: the promise of iPSC-derived neurons. Cell. 145 (6), 831-834 (2011).
- Pan, C., Kumar, C., Bohl, S., Klingmueller, U., Mann, M. Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Molecular & Cellular Proteomics. 8 (3), 443-450 (2009).
- Alge, C. S., Hauck, S. M., Priglinger, S. G., Kampik, A., Ueffing, M. Differential protein profiling of primary versus immortalized human RPE cells identifies expression patterns associated with cytoskeletal remodeling and cell survival. Journal of Proteome Research. 5 (4), 862-878 (2006).
- Yeo, Y., et al. Human Embryonic Stem Cell-Derived Neural Lineages as In Vitro Models for Screening the Neuroprotective Properties of Lignosus rhinocerus (Cooke) Ryvarden. BioMed Research International. 2019, (2019).
