1. Preparation of buffers and solutions for cell culturing
- Prepare collagen stock solution by dissolving 5 mg of collagen IV from human placenta in 50 mL of PBS overnight at 4 °C. Aliquot the stock solution into 5 mL portions and store at -20 °C.
- Prepare fibronectin stock solution by dissolving 5 mg of fibronectin in 5 mL of sterile water overnight. Store the fibronectin stocks in aliquots of 500 µL at -20 °C. When thawing, add PBS to a final volume of 50 mL to prepare the work solution and store it at 4 °C.
- Prepare Dulbecco's Modified Eagle Medium (DMEM) complete medium by adding 50 mL of fetal bovine serum (FBS), 5 mL of MEM nonessential amino acids and 5 mL of penicillin/streptomycin (0.1 g/L streptomycin sulfate and 100,000 U/L penicillin G sodium) to 500 mL of DMEM.
- Prepare 5 mg/mL heparin stock solution by dissolving heparin sodium salt in PBS and pass it through a 0.2 µm filter for sterilization. Store the stock solution at 4 °C.
- Prepare growth medium (GM) immediately before use; mix 10 mL of DMEM-comp and 250 µL of heparin stock solution per T75-flask.
2. Isolation of capillaries from fresh bovine brain
NOTE: Bovine brain capillaries are isolated and cultured as previously described (Helms et al.10).
- Collect brains from calves, no older than 12 months of age, from a slaughterhouse and bring directly to the lab on ice.
- Remove the meninges and collect all gray matter from the brain using a scalpel. Identify the meninges as the film covering the brain and the gray matter by its gray color.
- Use a 40 mL Dounce tissue grinder to homogenize the gray matter in Dulbecco's Modified Eagle Medium (DMEM). Fill the slim part of the tissue grinder 1/5 with gray matter suspension and add DMEM until the slim part is filled.
- Separate the capillaries from free cells and smaller tissue pieces by filtration of the homogenate through a 160 µm nylon net filter. Flush the filters with DMEM-comp. Retrieve the capillaries and pool the suspensions into 50 mL centrifugation tubes.
- Resuspend the capillaries in DMEM-comp and add an enzyme mix of DNase I (170 U/mL), collagenase type III (200 U/mL) and trypsin (90 U/mL). Leave the suspension for 1 h in a 37 °C water bath for digestion of the capillaries.
- Run the suspension through a 200 µm mesh filter and resuspend in FBS with 10% dimethyl sulfoxide (DMSO). Freeze the capillaries overnight at -80 °C and move them to liquid nitrogen the day after for long term storage.
NOTE: The protocol can be paused here.
3. Seeding and culturing of bovine capillaries
- Day 0: Mix 0.7 mL of collagen IV stock with 6.3 mL of PBS. Add the solution to a T75-flask and leave the flask for 2 h at room temperature (RT) or leave it overnight at 4 °C.
- Remove the collagen solution from the flask and wash three times with PBS.
- Add 7 mL of fibronectin work solution and leave the flask for 30 min at RT. Then, remove the fibronectin solution and seed the capillaries immediately after.
- During the 30 min waiting time, thaw one vial of capillaries in a 37 °C water bath.
- When the capillaries are thawed, transfer immediately to a centrifugation tube with 30 mL of DMEM-comp and centrifuge for 5 min at 500 x g and RT. Remove DMEM-comp from the tube and re-suspend the capillary-pellet in 10 mL of fresh DMEM-comp.
- Transfer the 10 mL suspension to the coated T75-flask and leave the capillaries to adhere to the bottom of the flask for 4-6 h in a 37 °C incubator at 10% CO2.
NOTE: The cell growth rate is higher at 10% CO2 rather than the conventional 5% CO2. - After 4-6 h of incubation inspect the flask under a light microscope. Fractions of capillaries should now be attached to the bottom of the flask (Figure 1, day 0).
- Prepare GM and aspirate the DMEN-comp medium very careful from the capillaries and replace it with 10 mL of freshly made GM.
- Day 2: Remove GM from the capillaries and replace with 10 mL of freshly made GM. Cellular outgrowth from the capillaries should be visible under a light microscope at this point (Figure 1, day 2-3).
4. Isolation of primary pericytes from bovine brain capillaries
- Day 4: Inspect the capillaries under a light microscope.
NOTE: The flask should now be approximately 60-70% confluent to provide an appropriate amount of pericytes (Figure 1, day 4). If this is not the case; replace the GM with 10 mL of fresh medium and leave the flask in the incubator for another day. - Aspirate the medium and wash the cells gently in PBS.
- Add 2 mL of thawed Trypsin-EDTA for endothelial cells and leave the flask in the incubator for 1-3 min. Take out the flask frequently and observe with the microscope during this time period.
NOTE: The endothelial cells should round up and detach from the flask; pericytes should be visible as cells with a "ghost"-morphology and still be attached to the surface of the flask. This is a tricky and important step. It is essential to remove most endothelial cells to avoid contamination of the pericyte monoculture, but prolonged trypsinization can also detach the pericytes. The trypsinization time can vary slightly from time to time, and it is therefore of utmost importance to observe the flask frequently with the microscope during the treatment. - Gently tap the flask, when the endothelial cells have started to round up, to detach the loosened endothelial cells.
- To stop the trypsinization, add 10 mL of DMEM-comp to the flask. Flush the flask carefully a few times with the medium to remove the endothelial cells. Aspirate the endothelial cell suspension from the flask. The endothelial cells can now be used for other purposes.
- Add 10 mL of DMEM-comp to the flask. Look under the light microscope to assure the pericytes are still present and attached to the bottom. Put the flask back into the incubator to allow the pericyte-enriched culture to grow.
NOTE: It is important to observe the culture during the following days. If there is still a fair amount of endothelial cells growing another trypsin-treatment can be performed. - Allow the pericyte monoculture to grow with change of DMEM-comp. medium every second day. Check the growth of the cells under the light microscope (Figure 1, day 5-8).
5. Generation and storage of a monoculture of primary bovine pericytes
- Day 8-9: Inspect the capillaries under a light microscope
NOTE: The pericytes should now have reached 70-80% confluency and grow in islands in the flask (Figure 1, day 9). If the confluency of the pericytes is less than 70%, allow the cells to grow for another day. The pericytes will not form a complete monolayer as the endothelial cells would. - Aspirate DMEM-comp and wash the pericytes with 7 mL of PBS.
- Add 2 mL of trypsin-EDTA to the flask and leave it in the incubator for 2-3 min. Place the flask frequently under the light microscope to observe when the pericytes round up and detach from the flask. When the pericytes have started to round up, the flask can be gently tapped to detach the cells.
- Gently tap the flask, when the pericytes have started to round up, to detach the cells.
- Add 10 mL of DMEM-comp to the flask to stop the trypsinization process. Flush the flask a few times with the medium to help detach the last pericytes.
- Transfer the 12 mL cell suspension to a 50 mL centrifugation tube and fill up to 30 mL with DMEM-comp.
- Centrifuge the cell suspension for 5 min at 500 x g and RT. Aspirate the DMEM-comp. carefully without touching the cell pellet. Resuspend the cell pellet in 3 mL of FBS with 10% DMSO.
- Transfer the cell suspension into cryovials; add 1 mL to each, so there will be a total of 3 vials per T75-flask of pericytes. Freeze the pericytes at -80 °C overnight and move them to liquid nitrogen the day after for long term storage.
NOTE: Cells may be counted before freezing for a later estimate of survival percentage. The protocol can be paused here.
6. Setting up a pericyte monoculture for experiments
- Coat a T75-flask with collagen IV and fibronectin using the same procedure as mentioned in section 3.1-3.4.
- While the flask is being coated with fibronectin, thaw one vial of pericytes in a 37 °C water bath.
- Transfer the now thawed pericytes from the cryovial to a centrifugation tube with 30 mL of DMEM-comp. Centrifuge the cell suspension for 5 min at 500 x g, RT.
- Carefully aspirate the medium, leaving the cell pellet at the bottom of the tube. Re-suspend the pellet in 10 mL DMEM-comp.
- Collect and transfer the cell suspension to the coated flask. Leave the flask with pericytes to grow in a 37 °C incubator at 10% CO2.
- Every second day, refresh the medium with 10 mL of fresh DMEM-comp.
NOTE: After 5 days of growth, the pericytes should have reached approximately 80% confluency. If the confluency is less, leave the cells to grow for another day or two. The cells should now be ready for seeding for further experiments.
7. Seeding of pericytes in a coated 96-well plate
- Dilute collagen IV as described in step 3.1. Add 100 µL to each well in a 96-well plate and incubate for 2 h at RT or overnight at 4 °C.
- Aspirate the solution and wash the wells three times with PBS.
- Add 100 µL of diluted fibronectin to each well and incubate at RT for 30 min. Remove the fibronectin solution and use the plate immediately.
NOTE: Depending on how well the pericyte batch is growing, there should be enough cells for seeding two plates. - Take out the pericytes from the incubator and aspirate the medium. Wash the cells with PBS.
- Add 2 mL of trypsin-EDTA to the pericytes and follow same procedure as in step 5.3-5.6.
- Aspirate the medium, without harming the cell pellet and re-suspend the pellet in 1 mL of fresh DMEM-comp.
- Take out 12 µL of cell suspension and add to a counting chamber. Under the light microscope, count at least 3 of 3×3 grids and use the average cell count per grid.
- Use the equation below to calculate the volume of cell suspension that should be added to each well to seed 10.000 cells per well, in the 96-well plate.
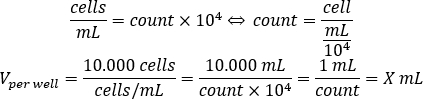
- Add DMEM-comp and the calculated volume of cell suspension in each well to reach a final volume of 200 µL.
- Place the 96-well plate in a 37 °C incubator at 10% CO2. Leave the cells to grow for 4 days with a change of medium after 2 days.
8. Preparation of buffers and solutions for Ca2+-imaging
- Autoclave coverslip cell chambers and coverslips.
- Assay buffer: Add 1.19 g of HEPES to 500 mL of HBSS buffer for a final concentration of 10 mM HEPES. Adjust the pH to 7.4.
- Prepare 20% (w/v) Pluronic F127 + 1% (v/v) polyethoxylated castor oil stock solution by dissolving 0.5 g of Pluronic F127 solution in 2.5 mL of anhydrous DMSO in a glass vial. Heat to 40 °C for approximately 30 min or until dissolved and vortex. Add 25 µL of polyethoxylated castor oil and store at RT. Do not freeze.
- Prepare 2 mM Fura-2 AM stock by dissolving 1 mg in 500 µL of anhydrous DMSO. Store in aliquots of 20 µL at -20 °C protected from light.
- Prepare 5 µM Fura-2 AM loading solution by mixing 20 µL of 20% Pluronic F-127 + 1% polyethoxylated castor oil stock solution with the 20 µL of 2 mM Fura-2 AM aliquot. Add 500 µL of assay buffer and vortex. Add assay buffer to a final volume of 8 mL. The solution should be prepared immediately before use and protected from light.
- Prepare 4 mM Cal-520 AM by dissolving 1 mg in 226.7 µL of anhydrous DMSO. Store in aliquots of 20 µL at -20 °C protected from light.
- Prepare 20 µM Cal-520 AM loading solution by mixing 20 µL of 20% Pluronic F-127 + 1% polyethoxylated castor oil stock solution with the 20 µL 4 mM Cal-520 aliquot. Add 500 µL of assay buffer and vortex. Add assay buffer to a final volume of 4 mL. The solution should be prepared immediately before use and protected from light.
9. Loading of pericytes with Fura-2 AM calcium indicator dye in a plate-reader setup
NOTE: All solutions should be at RT before the experiment starts.
- Take out the 96-well plate with cells from the incubator and aspirate the medium from the wells. Wash the cells twice with assay buffer.
- Add 100 µL of loading solution to each well and wrap the plate with tinfoil, to avoid photo bleaching. Incubate for 45 min with 30 rpm shaking at RT.
NOTE: Do not load Fura-2 AM at 37 °C, as it may load internal compartments. Remember to leave wells with cells in assay buffer instead of loading buffer; these are the "blanks" used for measuring background fluorescence. - Aspirate the loading buffer and wash the cells with assay buffer twice. Add 100 µL of fresh assay buffer and leave the cells to incubate for 30 min at RT; this allows for continuous cleavage of the AM-ester and thereby trapping Fura-2 AM inside the cells.
- Prior to the Ca2+-imaging, wash and replace the buffer with 100 µL of fresh assay buffer.
10. Well-plate fluorescence reading of pericytes in a plate-reader setup
- Set the temperature of the plate reader to 37 °C and transfer the 96-well plate with cells to the sample plate position. Place the reagent plate with agonist at the reagent plate position.
- Start by measuring loading of the cells to ensure equal loading of Fura-2 AM in all wells.
- Perform the measurements with excitation fluorescence wavelength at 340 nm/380 nm and the emission wavelength at 510 nm. Add 50 µL of agonist at speed 150 µL/s from the reagent plate to each well with cells in the sample plate position.
- Save the data and export as xlsx files for further analysis. Figure 2 shows the cytosolic Ca2+-response measured as the ratio between the two excitation wavelengths over time, where background fluorescence is subtracted.
NOTE: The plate-reader need to be a dual microplate reader with room for a "cell tray" and a "sample tray" and an integrated pipettor system.
11. Seeding of pericytes in a coated cell chamber for live imaging
NOTE: Coverslips may also be placed in the bottom of culture wells, coated and seeded with pericytes as described above, and then mounted in the chamber prior to experiments.
- Mount a coverslip into the cell chamber and make it tight to avoid leakiness.
- Dilute collagen IV as described in step 3.1. Add 500 µL to each cell chamber and incubate for 2 h at RT or overnight at 4 °C.
- Aspirate the collagen solution and wash the chambers three times with 500 µL of PBS.
- Add 500 µL of diluted fibronectin to each well and incubate at RT for 30 min. Remove the fibronectin solution and use the cell chamber straight afterwards.
- In the meantime, take out the flask with confluent pericytes and wash with 7 mL of PBS.
- Add 2 mL of trypsin-EDTA to the pericytes and follow same procedure as in step 5.3-5.6.
- Proceed by following the same steps as in step 8.6-8.7.
- Use the equation below to calculate the volume of cell suspension, which should be added to each chamber to seed 90.000 cells per chamber.
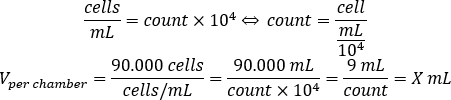
- Add DMEM-comp and the calculated volume of cell suspension in each chamber to reach a final volume of 500 µL.
- Place the cell chambers in the incubator at 37 °C, 10% CO2. Leave the cells to grow for 6 days (or until confluent).
NOTE: The pericytes grow slower on glass-slides compared to plastic; more days of growth are necessary.
12. Loading of pericytes with Cal-520 AM calcium indicator dye for live imaging
NOTE: All solutions should be at RT before the experiment starts.
- Prepare the 20 µM Cal-520 AM loading buffer: Mix 20 µL of 20% Pluronic F-127 + 1% polyethoxylated castor oil stock solution with the 20 µL 4 mM Cal-520 aliquot. Add 500 of µL assay buffer and vortex. Add assay buffer to a final volume of 4 mL. The solution should be prepared immediately before use and protected from light.
NOTE: Protect solutions containing Cal-520 AM from light exposure. - Take out the cell chambers from the incubator and aspirate the medium. Wash the cells twice with assay buffer.
- Add 500 µL of loading buffer to each chamber and incubate at RT for 45 min.
- Aspirate the loading buffer and wash the cells twice with assay buffer.
- Add 500 µL of fresh assay buffer to each chamber and incubate for 30 min at RT to allow cleavage of the AM-ester.
- Replace the buffer with 500 µL of fresh assay buffer before performing the live imaging at a confocal microscope.
13. Live imaging of intracellular Ca2+-levels
NOTE: A variety of microscope types can be used for the imaging. Upright or inverted conventional fluorescence microscopes, as well as upright or inverted confocal laser scanning microscopes with appropriate excitation source (488 nm) and emission filters (510-520 nm) can be used. Objectives should be suited for fluorescence and be of a high quality and with high numerical aperture (NA).
- Mount the cell chamber on the stage of the confocal microscope as gentle as possible, in order to avoid disturbance of the cells.
- Select an excitation wavelength of 488 nm, emission at 515 nm, sequential image acquisition with 5 second intervals, an XY image size of 512 x 512 pixels and measure for 2 min to measure baseline calcium signals.
- Add 3 µL of 100 mM ATP to the cell chamber with a pipette, and continue the sequential image acquisition. Perform the addition slowly and gently to not disturb the preparation and move the cells out of focus.
- Observe the degree of changes and increase the time interval over time as needed for approximately 18 min until no further morphological change is noted (Figure 3).
- Save time-lapse images and export them as TIFF and/or AVI files for further analysis.
NOTE: One vial of pericytes should give enough cells for seeding in 1-2 96-well plates and several coverslips, meaning you can prepare cells for both types of calcium-measurements.
Bovine brain capillaries were isolated from fresh brain tissue and Figure 1 presents the capillary seeding and cellular outgrowth over days and subsequent purification of pericytes. The capillaries are fully attached to the flask at day 1 and on day 2 endothelial sprouting has become visible (Figure 1, day 2). After 4 days, the cellular outgrowth is highly distinctive (Figure 1, day 4a) and the endothelial cells are removed by gentle trypsinization as according to the described protocol. Remnants of the capillaries can be present after the trypsinization, but will disappear from the flask in the following days (Figure 1, day 4b-6). After removal of the endothelial-layer, pericytes are easily detected with morphology distinct from the endothelial cells. The pericytes present finger-like processes that attach strongly to the flask (Figure 1, day 4b). Subsequently, pericytes are allowed to grow until confluency (Figure 1, day 4b-9) and on day 9, the pericytes have reached approximately 80% confluency and grow in islands. This is in contrast to the endothelial cells that do create a contact inhibited monolayer observed at day 4.
ATP is a well-known endogenous inducer of intracellular Ca2+-signaling11 and was used as an extracellular stimulant to induce cytosolic changes in Ca2+-levels in pericytes. Addition of ATP to the Fura-2 loaded pericytes, as according to the described protocol, resulted in an increase in cytosolic Ca2+-levels measured as the fluorescent ratio as shown in Figure 2. The ATP-induced response occurs immediately after addition of ATP to the pericytes and declines slowly over the measured time period.
Using this protocol for real-time confocal imaging of intracellular Ca2+-responses, pericytes were seeded on coated coverslips, loaded with Cal-520 AM and placed at the confocal microscope. Figure 3 (0 s) shows the pericytes with baseline levels of fluorescence prior to treatment. During live-recording, ATP is added to the pericytes and a strong intracellular Ca2+-response is evident shortly after (64 s). Soon after, the cytosolic Ca2+ compartmentalizes in the cells and a reduction in cell area is visible (189 s). At 300 s. post start of the recordings, the cell area is heavily reduced and the Ca2+-signal has declined to intensity close to the baseline fluorescence.
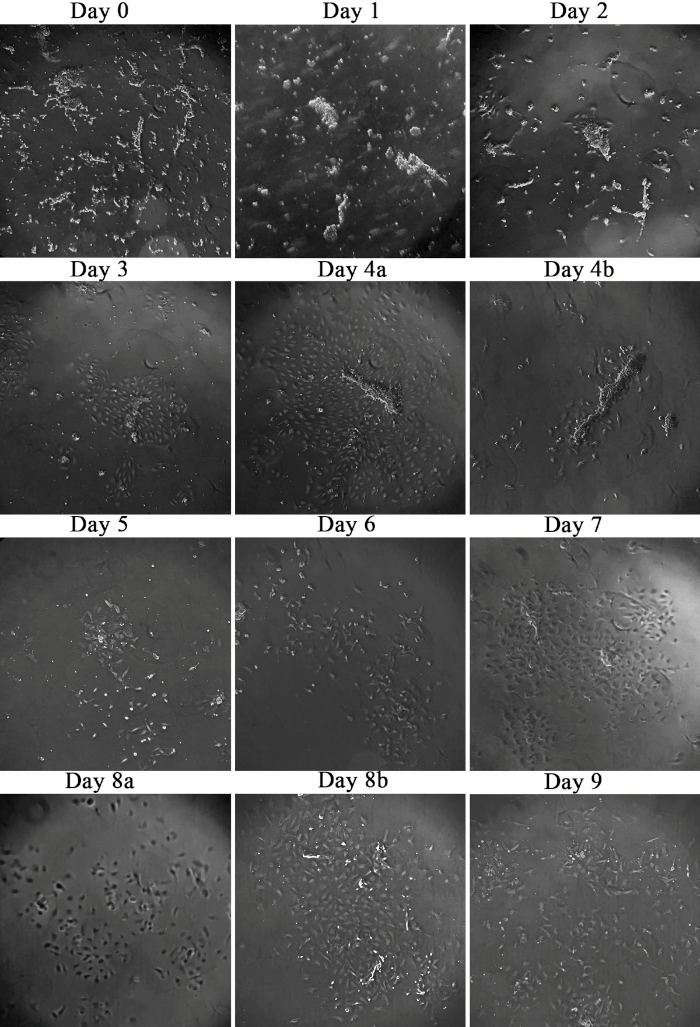
Figure 1: Culturing of capillaries and isolation of pericytes. Capillaries have been isolated from fresh bovine brain and seeded in culture flasks on day 0. Outgrowth from the bovine brain capillaries and the following isolation of pericytes were followed over days with a light microscope. Day 4a shows the endothelial cell growth prior to treatment with trypsin to remove endothelial cells and day 4b shows the remnants immediately after the treatment. Day 8a shows the focus plane, where any capillary remnants would be visible, whereas day 8b are focused on the plane where the growth of the pericytes is visible. Please click here to view a larger version of this figure.
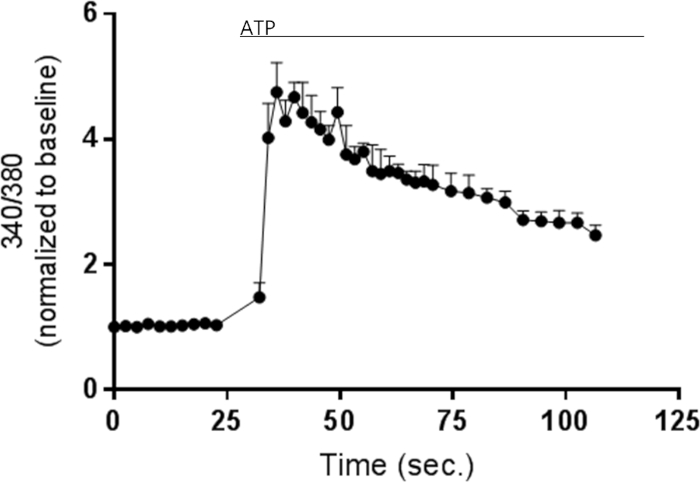
Figure 2: Representative example of intracellular calcium-measurement using Fura-2 calcium indicator dye. Primary pericytes have been seeded in 96 well plates and loaded with Fura-2 AM in order to visualize changes in intracellular calcium. 10 µM ATP is added to the pericytes at 30 s. and the cytosolic Ca2+-response is measured as the ratio between the two excitation wavelengths; 340 nm and 380 nm over time. Scale bars are defined as standard deviation (N=3, n=1). Please click here to view a larger version of this figure.
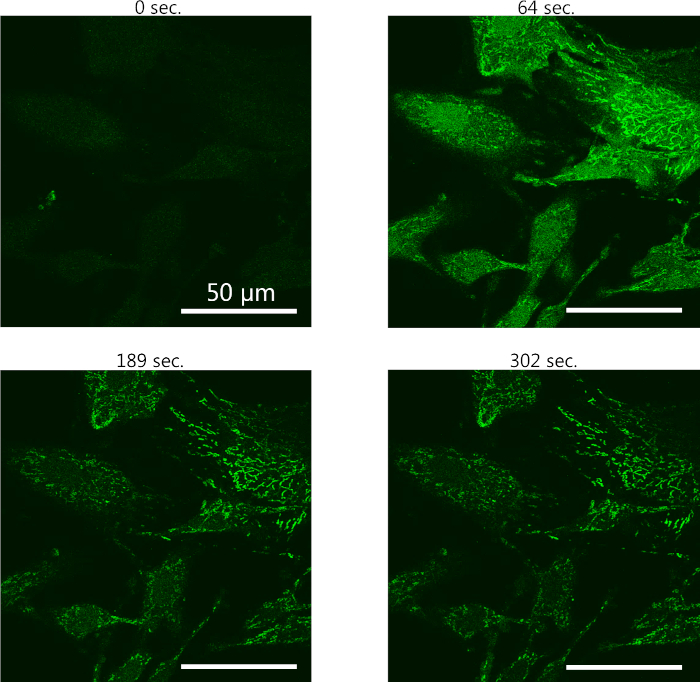
Figure 3: Representative example of intracellular calcium live-imaging using Cal-520 calcium indicator dye. Primary pericytes have been seeded in a cell chamber and loaded with Cal-520 in order to visualize changes in intracellular calcium and cell morphology. 600 µM ATP is added to the pericytes and snapshots from different time points during the live-imaging are presented here. Please click here to view a larger version of this figure.
